 and install Cvt2Mae on your computer and use it to
convert your array data to MAExplorer data format. Figure C.6.1 shows
Cvt2Mae array data converter.
and install Cvt2Mae on your computer and use it to
convert your array data to MAExplorer data format. Figure C.6.1 shows
Cvt2Mae array data converter.
|
Note: This appendix contains a "computerese" description on how
to use MAExplorer with your array data. The user-friendly "wizard"
tool |
MAExplorer requires a specification of array geometry and quantification information. These are defined in a configuration startup file. The startup file contains the initial list of hybridized samples to be loaded, and other parameters such as the name of the configuration file (if it is different from the default name). A stand-alone application causes the .mae startup file (or the PARAM list in the case of an applet) to be read when it is started. The configuration file contains various defaults. If any of these are specified in the configuration file, the override the built in default values. Values from the .mae startup or applet PARAMs will override the configuration file values. These configuration parameters may be overwritten by arguments in the stand-alone .mae startup files or PARAMs in the Applet startup specifications.
A few additional files are required and are defined in the configuration file. These include: a Gene-In-Plate-Order or GIPO file; a samples database file listing names of the samples available for loading; and a gene class names file. An optional (but deprecated) extra array information file may be specified to access additional data about samples. Quantified hybridized sample array spot data (Quant files) from each array is put into a separate data file. Note that all data files are tab-delimited files such as may be generated with Excel, relational databases or directly from array spot quantification software.
Hybridized sample arrays must be scanned and then spots quantified using other software. MAExplorer does not do spot quantification from scanned image files. However, MAExplorer can use spot data from a variety of array image quantification programs that generate tab-delimited data files. The data needs to be converted to the MAExplorer schema described in this Appendix.
The derivation of quantified spot data files from hybridized sample arrays is discussed later in this section as are in the quant file data format.
The configuration file is created once for each new array GIPO geometry and database of hybridized samples. It is independent of the number of samples. Configuration parameters include array geometry (# of grids, # of duplicate spots/gene, etc), whether the data is intensity or ratio data (e.g. Cy3/Cy5), etc. The configuration file may also include labeling, quantification dynamic range, default analysis thresholds, mapping of used data file table-field names to expected MAExplorer names for the GIPO and quantification files, additional database-specific pull-down menu plugins, names of gene sets and sample condition lists, etc.
The GIPO file is independent of the number of array samples and
describes the mapping between spot position in an array and its gene
identification as well as corresponding data such as original plate
number, row and column; UniGene ID, GenBank ID, dbEST ID, etc. These
files will be described in more detail including how one can create
the necessary database files that MAExplorer requires for use with
various types of microarray data.
Figure C.1 Directory structure of stand-alone databases required by
MAExplorer. The "/Config", "/Quant", and "/MAE" directories are
required. The /MAE directory is only used with the stand-alone version
with .mae files, not
for the applet. [When used with an applet, the main path is the path
of the download JAR file and .mae files are not used.] The "/Report",
and "/State" directories are created by MAExplorer as needed and the
user need not create them prior to running MAExplorer. The text
reports and plot GIF images are saved in the /Report folder when you
"Save" a report or plot. When you "Save" the current database session
(File | Databases | Save ...), the gene sets and sample lists are
saved in the /State folder for use when you restart MAExplorer on the
.mae startup file. The optional "/Cache" directory is only used (and
then, only optionally) when downloading data from a Web server. The
optional "/Image" directory is only used in there are JPEG images of
the arrays provided and their resolution and alignment must correspond
to the (X,Y) spot data in the Quant files. The "/Plugins" directory is
where the
MAEPlugins packaged with MAExplorer are normally kept and where
MAExplorer looks when you attempt to load a plugin. Since you can
browse your file system, they do not have to appear here.
Sample MGAP database configuration, quantification data and startup
files are available for use as examples with which to make your own
files or for inspection.
Similarly, when the entire database is saved (File | Databases |
SaveAs ...DB) into a .mae startup file, the set of gene set files are
saved as ".cbs" files and the set of condition list files are saved as
".hbl" files in the "State" subdirectory. These are automatically
reloaded into MAExplorer when the .mae startup file is used to restart
MAExplorer.
If your array data has JPEG or similar images of the original arrays,
the should be saved in the "Images" directory. For example, the
NCI-CIT mAdb database server allows you to download sampled images for
your data in an "Images" subdirectory at the same time you download
the other MAExplorer data files. The images can then be used by
various MAEPlugin programs. If your quantified data converted to
.quant files has (X,Y) coordinates corresponding to spots in these
images, then you may be able to use the Montage MAEPlugin to show
where the current spots are in sub-regions of all of the input
images. This plugin will be available on the MAEPlugin Web site when
we release the MAEPlugin facility for Beta-testing.
For a specific database (db), make sure the names of the configuration
files in /Config directory are entered in the
MaExplorerConfig-db.txt file for that database. You may have multiple
databases in the same /Config, /Quant and
/MAE directories if the file names do not conflict. The trick
is to have the .mae startup file in the /MAE
directory point to the specific configFile to be used. Since
MAExplorer reads the MaExplorerConfig-db.txt file when it first starts
up, it discovers the names of the other database files. If there is no
name conflicts, then there is no problem mixing data.
Each spot data (.quant) sample file has a name which must be entered
in the Database_File field of the Samples-db.txt row entry
for a new sample. The Sample_ID field is a descriptive name
of that sample.
Often GIPO files supplied by array vendors have additional fields not
currently used by MAExplorer. You can leave them in (they will be
ignored) or take them out (loading a database is faster).
If the field headings in the various user's tables are not the same
as that required by MAExplorer, you can easily fix this by adding
(Table,Field) mapping entries to your version of the
MaExplorerConfig-db.txt file (see mapTF
for examples).
Note that the optional Menu_Source_Name entry in the
Samples-db.txt file specifies the sub-menu, if any, that the sample
will appear in the Samples menu By Source sub-menu.
If the optional extra sample information file is used, then make sure
the sample names and database file names are the same, and that there
are corresponding rows in each table.
A typical sample database table might look like:
Directory (i.e. folder) structure of stand-alone databases
When running as a stand-alone application, MAExplorer assumes that
data from a local computer has a specific directory structure. The
required and optional directories (also called "folders" on some
operating systems) and files they contain are diagramed here from a
database project directory in your file system. The notation
"/folder-name" indicates that "folder-name" is a folder inside of the
project.
(specific database directories and files they contain)
/ Cache
/ (copies of any data files saved from Web DB access)
/ Config
/ MaExplorerConfig.txt
/ SamplesDB.txt
/ GIPO-db.txt
/ MAE
/ (set of startup database files).mae
/ Images
/ (set of original or sampled array .jpg images) (optional)
/ Plugins
/ (optional set of .jar or .class MAEPlugin files)
/ Quant
/ (set of spot quantified data files).quant
/ Report
/ (set of .txt and .gif report files generated using SaveAs
/ State
/ (set of gene set files).cbs and
/ (set of condition list files).hbl generated using Save DB
Examples of some of the database files required by MAExplorer
These could be used as examples that could be used in creating your
own database files. When the MAExplorer converter tool, Cvt2Mae, is
released it will eliminate the need for manually editing your database
files.
In addition, examples of the (Config/, Quant/ and MAE/) files
needed for various types of arrays are available at:
Additional directories used at run-time
When running MAExplorer as a stand-alone application, you may save
data on the disk Text reports and plot graphics windows are saved as
".txt" text and ".gif" image files when the user uses the "SaveAs"
button in the respective popup windows. These files are saved in the
"Report" subdirectory.Tools for automating the construction a local stand-alone database
Software tools for aiding the construction of local stand-alone
databases from vendor supplied GIPOs and spot quantification files are
not available at this time, but will be made available in the future.
Manually constructing a local stand-alone database
Although the Cvt2Mae converter tool can convert many files, you could
alternatively build these files manually. We suggest using Excel or
your favorite RDBMS system to manipulate the data. At the end, save
the data into files with tab-delimited fields with the above file
extensions (i.e. .txt, .quant, .mae). The layout of these files and
what is optional and what is not is described in detail (maybe too
much!) below. You could use an ASCII file text editor instead of Excel
(such as Wordpad, Emacs, etc.) - but be careful not to add or
delete tabs since this will destroy the integrity of the database
tables. Be consistent in your file names; avoid spaces; use ASCII
characters in file names that are system independent (i.e. A-Z,
a-z,0-9, "-", "+", "_"); Use either "-" or "_" or both. C.1 Creating quantified spot data files from hybridized sample arrays
Quantified spot data from images scanned from hybridized sample arrays
may be created using a variety of software programs. Discussion of
these is beyond the scope of this manual. However, several of these
including Pathways 2.01, ImageQuant-NT, and others generate
tab-delimited text files. These files may be used directly as the
quantified spot files required by MAExplorer, or simplified first (by
removing unused or redundant data fields). Typically, the files are
named (or renamed) to that of the sample to distinguish them from each
other and a .quant file extension assigned instead of the
.txt file extension. Other programs generate tab-delimited
files that could be mapped to our .quant file formats. (For example,
the NCI/CIT mAdb system
generates such a mapping for GenePix(TM), and ScanArray
formated data.)
C.1.1 Color and prefix notation for the following tables:
(req), (opt), (future)
The following tables list parameters and some typical values
that might be included in the configuration and quantification files.
These examples illustrate the variety of parameters or fields with
examples of values that might be used. Required parameters are
in black with "(req)" prefix. Optional parameters are
indicated in blue with a "(opt)"
prefix. Optional parameters are not normally specified and are
generated in the .mae file when you save the state of a data
exploration. Parameters that might be used with Cy3/Cy5 ratio data
are indicated in magenta with a "(ratio)"
prefix. Future options not currently used are indicated in
green with a "(future)" prefix. Alternative
options are indicated in red with an "(alt)"
prefix.
C.2 Table of samples that can be loaded into MAExplorer
The samples available to be analyzed in a database are listed in a
samples database table. This lists all samples that
could be loaded. The user will then select a subset of these to
be analyzed. The selection is done either in preset Web startup pages,
or with the stand-alone application .mae startup files, or at
run-time by selecting new entries from the Samples pull-down
menus. Extra information may be provided to MAExplorer for each sample
through this table and will be available for the Sample Array report in
Section 2.4.6.1.
Sample_ID Project Database_File control 1 breastCancer control1 control 2 breastCancer control2 control 3 breastCancer control3 tumor 1 breastCancer tumor1 tumor 2 breastCancer tumor2 tumor 3 breastCancer tumor3 |
You may optionally include a Database_ID field. For example:
Sample_ID Project Database_File Database_ID control 1 breastCancer control1 270314 control 2 breastCancer control2 270315 control 3 breastCancer control3 270316 tumor 1 breastCancer tumor1 270317 tumor 2 breastCancer tumor2 270318 tumor 3 breastCancer tumor3 270319 |
The Database_ID may be useful if there are file length problems on some systems (i.e. MacOS 8-9), we offer the option of using the Database_ID as the file name for the .quant (Quant/ directory) and .jpg (Images/ directory) rather than the Database_File name. For example one could specify "Quant/270314.quant" and "/Images/270314.quant" rather than the default "Quant/control1.quant" and "/Images/control1.quant" names.
The Samples database table includes some required as well as optional fields (see Table C.2.1.1):
Table C.2.1 List of Samples data file table fields. The Samples table lists hybridized samples that are accessible to the user and may be loaded into a database session if they wish. (See Section C.1.1 for option notation.)
| Field | Description |
|---|---|
| (req) Sample_ID | descriptive name of the sample, free text. [Note: an older depricated name is "Membrane_ID"] |
| (req) Project | that the sample belongs. Used for login protection and grouping of samples |
| (req) Database_File | name of the .quant spot database file, no spaces. This is the file name for the sample. |
| (opt) DatabaseFileID | database file ID corresponding to Database_File and Sample_ID. For use with RDBMS Web databases (e.g. experiment id #). NOTE: if you are encoding auxillary data files using this identifier, e.g. sampled array images in the Images/ directory, then this field is required if you want to access those images. |
Table C.2.1.1 List of optional Samples data file table fields. These fields may be used for some additional operations. If they are not in the Samples DB table, then the operations will not be available. (See Section C.1.1 for option notation.)
| (opt) Menu_Source_Name | Sample SubMenu j that this sample belongs. You could use the word "Default" or leave out this entry if you do not want to use sub menus. |
| (opt)Orig_File_Name | if applicable. The original file name and sample name if the data was split out from a multiple hybridized sample file. |
| (opt)Strain | if applicable |
| (opt) Source | if applicable |
| (opt) Probe | if applicable |
| (opt) Stage | if applicable (eg, developmental stage, dose, time point, etc) |
| (opt) Login | (optional) TRUE if login required with a Web server else blank. This is used primarily with the Applet when interacting with a Web server |
| (opt) GeneCard_URL | GeneCard ID if applicable |
| (opt) Histology_URL | (e.g. MGAP) histology DB Web page if applicable |
| (opt) Model_URL | (e.g. MGAP) mouse model database Web page if applicable |
| (opt) BGLow | global low value of array background intensity |
| (opt) BGAvg | global average value of array background intensity |
| (opt) BGRms | global root-mean-square value of array background intensity |
Table C.2.1.2 List of optional Samples data file table fields. These fields are not currently used in any computations but are returned in the Sample Array report in Section 2.4.6.1.
| (opt) Contributor | name of researcher submitting the sample |
| (opt) Contrib_Institute | researcher's organization |
| (opt) Submission_Date | when submitted |
| (opt) Exposure | minutes or hours of radiolabel or fluorescent exposure |
| (opt) Sample_Nbr | internal sample number |
| (opt) FilterType | name of the array layout |
| (opt) FilterType_Description | additional description of array layout |
| (opt) Comments | details describing sample |
| (opt) Researcher | researcher performing the hybridization |
| (opt) SampleGrid | serial number of the array or grid or internal laboratory numbering. (Useful if reusing arrays etc) |
C.3 Quantified spot data file formats
MAExplorer has been designed to be able to read quantified spot data
from a variety of spot analysis software packages. So the data file
format is very flexible. Essentially, a data file contains one or
more spot intensity values per gene in each row of the data file. A
spot location is specified by a GIPO (field#, grid#, grid column#,
grid row#) 4-tuple with the field value optional. Note: a "grid" is
sometimes called a "block" or a "patch". If the field specification is
omitted and there are duplicate spots in multiple fields of grids,
then it is defined implicitly. In that case, the corresponding spot
intensity data for each field for a gene is specified as separate
columns going from left to right. The (grid#, column#, row#) part of
the specification may be encoded several ways: a) explicitly as
(grid#, column#, row#) or b) NAME_GRC.
|
The basic Quant spot data file table includes entries listed in Table C.3.1:
Table C.3.1 List of Quant data file table fields. This specifies the spot quantification data. There may be one or more spots, corresponding to the same gene, on each row. (See Section C.1.1 for option notation.)
| Field | Description |
|---|---|
| (opt) field | field for duplicate genes if using single 'RawIntensity' value/Row |
| (req) grid | grid name (either A,B,C,... or 1,2,3,... ) |
| (req) grid col | column with in a grid |
| (req) grid row | row within a grid |
| (opt+alt) NAME_GRC | (alternative specification of "grid, grid col, grid row"). |
| (req) RawIntensity1 | intensity value for field 1. Use this form if there is more than 1 intensity value/row. |
| (req) RawIntensity2 | intensity value for field 2 (required if it exists and for Cy3, Cy5 data) | (req+alt) RawIntensity | intensity value for field 1, if only one field used |
| (opt) Background1 | background intensity value for field 1 |
| (opt) Background2 | background intensity value for field 2 (if it exists for F1,F2 data or Cy3, Cy5 data) |
| (opt+alt) Background | background intensity value for field 1, if only one field used |
| (opt) QualCheck | quality check for data indicating "bad" spots or genes. Current codes are listed in the Table C.4.2 of QualCheck semantics |
| (opt) DetValue | spot data detection value quality. This could be the Affymetrix MAS5.0 "Detection p-value" or some other metric correlated with spot detection quality in the range of [0.0 : 1.0]. metrix |
Note: If NAME_GRC is specified (eg. for use with ImageQuant-NT data), then the explicit (grid, grow row, grid col) fields are not required. Note: For [G grids, R rows and C columns], this would cover a set of spots in the range [1,1,1] through [G,R,C].
Note: If Cy3/Cy5 double fluorescent labeling is used, then the RawIntensity1 and RawIntensity2 fields may be replaced with Cy3RI and Cy5RI names and the (RawIntensity1, RawIntensity2) fields mapped to (Cy3RI, Cy5RI) in the configuration file mapTF entries (table C.5.4 below). (See Section C.1.1 for option notation.)
| Field | Description |
|---|---|
| (req) Cy3RI | RawIntensity1 value for Cy3 |
| (req) Cy5RI | RawIntensity2 value for Cy5 |
| (opt) Cy3Bkgrd | Background1 value for Cy3 |
| (opt) Cy5Bkgrd | Background2 value for Cy5 |
| (opt) Cy3 | RawIntensity1 value for Cy3 |
| (opt) Cy5 | RawIntensity2 value for Cy5 |
Data is extracted from a table created from the gene-in-plate-order (GIPO) gene coordinate table. This links spots in a microarray to these Genomic "gene ID"s and gene names. This table may contain Clone ID, GenBank, dbEST, UniGene IDs, LocusID corresponding to these Master Gene IDs. An optional table of Clone IDs and Gene Classes the gene belongs to may also be defined.
A typical GIPO database table might look like:
Location grid grid col grid row plate plate row plate col Clone ID GenBankAcc GeneName . . . 39 A 2 15 2 1 3 1247601 AA763423 "Mus musculus A kinase anchor protein (AKAP-KL) mRNA, alternatively spliced isoform 1, complete cds" 40 A 2 16 2 1 4 1247553 AA763380 Mus musculus bodenin gene 41 A 2 17 2 1 5 1247865 AI465019 "Mouse beta-D-galactosidase fusion protein mRNA, complete cds" . . . |
The basic GIPO table includes the following fields:
| Field | Description |
|---|---|
| (opt) field | array field for duplicate genes |
| grid | array grid name (either A,B,C,... or 1,2,3,... ) |
| grid col | array column within a grid (either A,B,C,... or 1,2,3,... ) |
| grid row | array row within a grid (either A,B,C,... or 1,2,3,... ) |
| (opt+alt) NAME_GRC | alternative specification to "grid, grid col, grid row". It is generated by the Molecular Dynamics spot quantification software. |
| (opt) Master Gene ID | This is the master gene identifier used in MAExplorer. It must be one or more of the identifiers listed in Table C.4.3. One of these will be selected as the Master Gene ID (MID) |
| (req) Gene Name | Master Gene Name. The GeneName options are listed in Table C.4.1. These alternative GeneClasses are automatically recognized from the Gene Name. |
| (opt) plate | plate name for original gene. If this is not specified, it uses the grid value. |
| (opt) plate row | plate row name for original gene. If this is not specified, it uses the grid row value. |
| (opt) plate col | plate column name for original gene. If this is not specified, it uses the grid col value. |
| (opt) QualCheck | quality check for data indicating "bad" spots or genes. Current codes are listed in the Table C.4.2 below |
| Field | Description |
|---|---|
| (opt) GeneName | Gene name |
| (opt) Unigene cluster Name | alternative for GeneName if the latter is not specified. |
For example, if grid #, row# and column# are (8,12,11), then it codes it as
Automatic Gene Class naming based on Gene Name
Some Gene Classes are automatically recognized from
the Gene Name including:
Alternative Grid,Row,Column encoding scheme: NAME_GRC
Some quantification programs (e.g. Molecular Dynamics "ImageQuant-NT) specify
"grid, grid_col, grid_row" by a single symbol we denote NAME_GRC coded
as follows
GRID- grid#-Rrow#Ccol#
GRID- 8-R12C11
Table C.4.2 List of QualCheck codes and their semantics
The data filter "Filter by 'Good Spot data'" may be used in
eliminating bad spot data on a per-gene set basis. This uses the
"QualCheck" field in the quantified data table is present. It maps
either an 1) integer numeric code (see Appendix C of the Reference
Manual), 2) an alphabetic code (e.g. Affymetrix "Abs Call") of "P" (or
"G" or "T") to Good Spot, "A" (or "B" or "F") to Bad Spot, and "M" to
Marginal Spot, or 3) a continuous quality value. In this latter case,
QualCheck may be a continuous monotonically increasing floating point
value (e.g. 0.0 to 100.0, or 0.0 to 1.0, -100.0 to +100.0, etc.) in which case a "Spot
Quality" State threshold slider will popup when the filter is invoked.
Additional property value codes may be added in the future.
| Status | QualCheck value | Semantics |
|---|---|---|
| Good gene | 2 | the spot data is "Good" (some systems report this by a NULL quality measure). It has a good gene name. Alternatively, letter codes may be used "P", "G", "T". |
| Bad gene | 4 | the spot data is bad, a good gene name. |
| Bad spot | 8 | is a non-analyzable spot (eg. marker, or "Bad", "Not Found", "Empty". etc.) Alternatively, letter codes may be used "A", "B", "F". |
| Duplicate spot | 16 | is duplicate of another gene on array |
| Marginal spot | 256 | is a marginally quantified spot. Alternatively, letter codes may be used "M". |
| Field | Description |
|---|---|
| (opt) Location | alternate spot identifier. E.g., Affymetrix 'probe_set', or Incyte 'IncyteID', etc. This may be numeric or alphanumeric |
| (opt) Clone ID | I.M.A.G.E. consortium database clone ID. It may have a "IMAGE:" or "ATCC:" prefix |
| (opt) Unigene cluster ID | NCBI UniGene database ID |
| (opt) dbEST3' | NCBI dbEST database |
| (opt) dbEST5' | NCBI dbEST database |
| (opt) GenBankId | NCBI GenBank database |
| (opt) GenBankId3' | NCBI GenBank database |
| (opt) GenBankId5' | NCBI GenBank database |
| (opt) RefSeqID | NCBI RefSeq database |
| (opt) LocusID | NCBI LocusLink database |
| (opt) OMIMID | NCBI OMIM database |
| (opt) SwissProtID | Swiss-Prot database |
| Parameter | Value | DataType | Comments |
|---|---|---|---|
| (opt) GenomicMenu1 | GenBank | String | Name of the database. This will appear in the View menu |
| (opt) GenomicURL1 | http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=2&form=1&term= | String | URL to which one adds the 'GenomicIDreq' value |
| (opt) GenomicURLepilogue1 | String | epilogue of the URL if any | |
| (opt) GenomicIDreq1 | GBID | String | Name of the GenomicID required and that is specified in the GIPO file as one of its fields |
| (opt) GenomicMenu2 | UniGene | String | Name of the database. This will appear in the View menu |
| (opt) GenomicURL2 | http://www.ncbi.nlm.nih.gov/UniGene/query.cgi?ORG=Mm&CID= | String | URL to which one adds the 'GenomicIDreq' value |
| (opt) GenomicURLepilogue2 | String | epilogue of the URL if any | |
| (opt) GenomicIDreq2 | UID | String | Name of the GenomicID required and that is specified in the GIPO file as one of its fields |
We are developing tools for creating and editing the configuration file. In the mean time, edit the file with Excel and save the finished table as a tab-delimited text file with the name MaExplorerConfig.txt in the Config sub-directory) in the directory where your database is stored.
Table C.5 List of Configuration data file table fields.
| Parameter subset | Function of these parameters |
|---|---|
| 1. Array content & geometry | Describes the content and geometry of the arrays (required) |
| 2. Threshold defaults | Describes the threshold defaults (optional) |
| 3. Array database files | Describes the array specific database files (required) |
| 4. Table field mapping | Describes "mapTF" table,field mapping. This maps user defined names to names required by MAExplorer and is only required if the user names are different from the names MAExplorer expects. |
| 5. URL genomic databases | Describes base addresses of genomic Web DBs (optional). If you do not specify these, default values are supplied from the program. |
| 6. User menus | Describes user-specific menus (optional) |
The following sub-tables list the configuration parameters and some typical values that might be included. These examples illustrate the variety of parameter options with examples of values that might be used. Required entries are listed at the tops of the tables.
A typical MAExplorer minimal configuration database table might look like:
Parameter Value DataType Comments MAX_FIELDS 1 int # replicate grids/array MAX_GRIDS 2 int # grids/field MAX_GRID_COLS 38 int # columns/grid MAX_GRID_ROWS 27 int # rows/grid usePseudoXYcoords true boolean use pseudoarray XY coord image - no XY data gipoFile GIPO.txt File name of GIPO file from samplesDBfile SamplesDB.txt File name of Samples DB file dataBase demo String default name of project database dbSubset demo1 String default database subset name useRatioData true boolean treat duplicate(F1,F2) data as ratio (F1/F2) - i.e.Cy3/Cy5 EditDate Tue Aug 21 2000 String demo |
| Parameter | Value | DataType | Comments |
|---|---|---|---|
| (req) MAX_FIELDS | 2 | int | # duplicate grids (blocks, patch, etc.) of spots for each gene in the array (i.e. F1, F2, etc.). Note that Cy3 and Cy5 data for each spot count as one field. |
| (req) MAX_GRID_COLS | 24 | int | # cols/grid in the array |
| (req) MAX_GRID_ROWS | 9 | int | # rows/grid in the array |
| (req) MAX_GRIDS | 8 | int | # grids in the array |
| (opt) ignoreExtraFields | FALSE | boolean | if there are additional fields of data in the GIPO or .quant files, then ignore them. Only use the first rawIntensity field. Note: this option is not normally used. |
| (opt) reuseXYcoords | FALSE | boolean | Reuse XY coordinates from first sample for rest of the samples |
| (opt) SpotRadius | 7 | int | (2 to 20 pixels) 50 microns, scroller. Note: this should be set to about 4 or 5 for a 10000 gene DB. |
| (opt) swapRowsColumns | FALSE | boolean | set if swap rows and columns in the array (used with our particular Research Genetics arrays) |
| (opt) usePseudoXYcoords | FALSE | boolean | use pseudoarray XY coordinates image if there is no explicit no XY spot position data generated by the quantification software |
| (future) FIELD_LAYOUT | LtoR | String | fields are Left to Right |
| (future) FIELDS_ARE_NUMBERED | TRUE | boolean | Data files contain field number. Otherwise field is extrapolated |
| (future) GRID_LAYOUT | Horizontal | String | Grids are Left To Right in the array |
| (future) GRID_PER_ROW | 4 | int | # grids per row in each field of the array |
| Parameter | Value | DataType | Comments |
|---|---|---|---|
| (ratio) fluorescentLbl1 | Cy3 | String | name of dye for fluorescent label 1 |
| (ratio) fluorescentLbl2 | Cy5 | String | name of dye for fluorescent label 2 |
| (ratio) useRatioData | TRUE | boolean | set if data is Cy3/Cy5 ratio data otherwise it assumes intensity data for each spot |
| (opt+ratio) useRatioMedianCorrection | FALSE | boolean | when using ratio data mode (Cy3/Cy5), use ratio median correction as the default |
| (opt) useBackgroundCorrection | FALSE | boolean | use background correction as the default when startup |
| (future) useCy5/Cy3 | FALSE | boolean | compute Cy5/Cy3 ratios instead of Cy3/Cy5 ratios |
| Parameter | Value | DataType | Comments |
|---|---|---|---|
| (opt) calibDNAname | mouse genomic DNA | String | name for calibration DNA if available - replacing cloneID in the case where the clones are not yet in the I.M.A.G.E. database. The particular clone is located using the Plate(grid,row,col) reported when selecting the current gene. |
| (opt) classNameX | HP-X 'set' | String | default name of HP-X samples 'set' |
| (opt) classNameY | HP-Y 'set' | String | default name of HP-Y samples 'set' |
| (opt) dataBase | MGAP DB | String | name of the database project |
| (opt) dbSubset | Preg 13 vs Lact 1 | String | name of the subset of data from the database |
| (opt) geoPlatformID | GPL80 | String | name of the NCBI Gene Expression Omnibus (GEO) Platform Id |
| (opt) maAnalysisProgram | Research Genetics Pathways 2.01 | String | name of spot quantification program |
| (opt) yourPlateName | your plate | String | name of researcher's clones if available - used in the cloneID data field in the case where the clones are not yet in the I.M.A.G.E. database. The particular clone is located using the Plate(grid,row,col) reported when selecting the current gene. (See Table 2.4.1) |
| (opt) emptyWellName | empty wells | String | what you called empty wells if there are any in the database. (See Table 2.4.1) |
| (opt) EditDate | 06-19-00, Lemkin | String | comment why changed |
| Parameter | Value | DataType | Comments |
|---|---|---|---|
| (opt) gangSpotFlag | TRUE | boolean | set gang spot display on startup for database with duplicate spots |
| (opt) presentationViewFlag | FALSE | boolean | start MAExplorer with larger fonts and graphics symbols suitable for live presentations |
| (opt) showEGLflag | FALSE | boolean | show EGL genes on startup from previously saved database that had EGL genes selected. |
| (opt) showMouseOver | TRUE | boolean | show mouse-over info when move mouse in windows |
| (opt) useDichromasy | FALSE | boolean | use orange-blue else use red-green color scheme |
| (opt) viewFilteredSpotsFlag | TRUE | boolean | view Filtered spots the array pseudoimage. If it is off, it shows just the pseudoarray image without spots passing the filter or MAExplorer state information. |
Note that there are many other parameters reflecting the state of MAExplorer that are saved in the .mae startup file when doing a (File | Database | SaveAs...DB) operation. These are reviewed and set from the MAExplorer menus. These parameters are not listed here - although they could be used in setting up an initial .mae startup file.
| Parameter | Value | DataType | Comments |
|---|---|---|---|
| (opt) CanvasHorSize | 1100 | int | pixels, horizontal size of microarray image **DEPRICATED** |
| (opt) CanvasVertSize | 1100 | int | pixels, vertical size of microarray image **DEPRICATED** |
| (opt) fontFamily | SansSerif | String | default text font family. See Font Family for other fonts. Some fonts look better with some operating systems. |
| (opt) clusterDistThr | 10 | float | default cluster similarity threshold in [0.0 : 100.0], scroller |
| (opt) maxGenesReported | 50 | int | max # of genes in highest/lowest gene report |
| (opt) maxPreloadImages | 4 | int | max # HP samples to initially load |
| (opt) nbrOfClustersThr | 6 | int | default # clusters for K-means clustering |
| (opt) pValueThr | 0.2 | float | default p-value for statistical tests |
| (opt) spotCVthr | 0.25 | float | default spot Coefficient of Variation value |
| (opt) allowNegQuantDataFlag | FALSE | boolean | set if .quant file data has negative intensity values otherwise it clips the negative values to 0.0 |
| (opt) usePosQuantDataFlag | TRUE | boolean | Filter out genes where .quant file data has negative intensity values otherwise it uses the negative data |
| Parameter | Value | DataType | Comments |
|---|---|---|---|
| (req) gipoFile | GIPO-DB.txt | File | Composite Gene-In-Plate-Order (GIPO) file containing the spot print order, Clone-IDs, gene names, GenBank IDs, plate coordinates, etc. (See Appendix C.4) |
| (req) samplesDBfile | Samples-DB.txt | File | list of hybridized samples in the database. [Note: an older depricated name was "membranesDBfile"]. (See Appendix C.2) |
| (opt) quantFileExt | .quant | String | alternate quantification spot file name extension to use instead of ".quant". (You might set it to ".txt") (See Appendix C.3) |
[TableName],[MAE field name],[TableName],[User field name]
The following table fields may be mapped. Note: mapping is required only when the table field names of your data files are different than the internal MAExplorer table field names.
The following is an example of some of the parameters that might be added to the Configuration file to perform field name mappings. Note: these mappings are only required if the data field names are non-standard. This shows some typical field name mappings. It will not be the same for your data. (See Section C.1.1 for option notation.)
| Parameter | Value | DataType | Comments |
|---|---|---|---|
| (opt) mapTF | GipoTable,grid,GipoTable,SA | String | GIPO table grid name (numbers or letters) |
| (opt) mapTF | GipoTable,grid row,GipoTable,R | String | GIPO table row of grid name (numbers or letters) |
| (opt) mapTF | GipoTable,grid col,GipoTable,C | String | GIPO table column of grid name (numbers or letters) |
| (opt) mapTF | GipoTable,plate,GipoTable,RG Pl | String | GIPO table plate where clone came from |
| (opt) mapTF | GipoTable,plate row,GipoTable,RG row | String | GIPO table row of plate where clone came from |
| (opt) mapTF | GipoTable,plate col,GipoTable,RG col | String | GIPO table column of plate where clone came from |
| (opt) mapTF | GipoTable,Clone ID,GipoTable,Clone id | String | GIPO name of Clone ID |
| (opt) mapTF | GipoTable,GeneName,GipoTable,Gene name | String | GIPO table map gene name |
| (opt) mapTF | GipoTable,Unigene cluster ID,GipoTable,ucid | String | GIPO table UniGene cluster id (if available) |
| (opt) mapTF | Unigene cluster name,GipoTable,ucn | String | GIPO table UniGene cluster name (if available) |
| (opt) mapTF | GipoTable,GenBank 3',GipoTable,gb3' | String | GIPO table GenBank 3' id (if available) |
| (opt) mapTF | GipoTable,GenBank 5',GipoTable,gb5' | String | GIPO table GenBank 5' id (if available) |
| (opt) mapTF | GipoTable,dbEST 3',GipoTable,est3' | String | GIPO table dbEST 3' id (if available) |
| (opt) mapTF | GipoTable,dbEST 5',GipoTable,est5' | String | GIPO table dbEST 5' id (if available) |
| (opt) mapTF | QuantTable,grid,QuantTable,SA | String | Quant table array grid name (numbers or letters) |
| (opt) mapTF | QuantTable,grid row,QuantTable,R | String | Quant table row of grid name (numbers or letters) |
| (opt) mapTF | QuantTable,grid col,QuantTable,C | String | Quant table column of grid name (numbers or letters) |
| (opt) mapTF | QuantTable,RawIntensity,QuantTable,Intensity | String | Quant table RawIntensity data |
| (opt) mapTF | QuantTable,Background,QuantTable,BkgrdIntens | String | Quant table background intensity |
| (opt) mapTF | QuantTable,RawIntensity1,QuantTable,Cy3RI | String | Quant table RawIntensity1 Cy3 data |
| (opt) mapTF | QuantTable,RawIntensity2,QuantTable,Cy5RI | String | Quant table RawIntensity2 Cy5 data |
| (opt) mapTF | QuantTable,Background1,QuantTable,BkgrdCy3RI | String | Quant table background intensity for Cy3 |
| (opt) mapTF | QuantTable,Background2,QuantTable,BkgrdCy5RI | String | Quant table background intensity for Cy5 |
| Parameter | Value | DataType | Comments |
|---|---|---|---|
| (opt) dbEstURL | http://www.ncbi.nlm.nih.gov/irx/cgi-bin/birx_doc? dbest+ |
String | NCBI dbEst server by dbEST ID. You may use an alternative server. |
| (opt) GenBankAccURL | http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Search&db=Nucleotide&term= |
String | NCBI GenBank server by GenBankAcc ID. You may use an alternative server. |
| (opt) GenBankCloneURL | http://www.ncbi.nlm.nih.gov/irx/cgi-bin/submit_form_query? TITLE=dbEST+Retrieval+Output&INPUTS=1& BRACKETS=NONE&ADDFLAGS=-b&DB=dbest& NDOCS=10&Q1= |
String | NCBI GenBank entry by Clone_ID server. You may use an alternative server. |
| (opt) GenBankCloneURLepilogue | [clin] | String | Epilog added after Clone_ID. You may use an alternative server. |
| (opt) IMAGE2GenBankURL | http://nciarray.nci.nih.gov/cgi-bin/UG_query.cgi? ORG=Mm&ACC=IMAGE: |
String | lookup GenBank from CloneID server. You may use an alternative Image to GenBank server. The "ORG=Mm" should be changed to reflect the proper species, eg. "ORG=Hs" for human, etc. |
| (opt) IMAGE2GIDURL | http://nciarray.nci.nih.gov/cgi-bin/UG_query.cgi? ORG=Mm&GID=IMAGE: |
String | NCI/CIT lookup GenBank GID from CloneID server. You may use an alternative CloneID to GenBank GID server. The "ORG=Mm" should be changed to reflect the proper species, eg. "ORG=Hs" for human, etc. |
| (opt) IMAGE2unigeneURL | http://nciarray.nci.nih.gov/cgi-bin/UG_query.cgi? ORG=Mm&CLONE=IMAGE: |
String | NCI/CIT lookup UNIGENE from CloneID server. You may use an alternative CloneID to UniGene server. The "ORG=Mm" should be changed to reflect the proper species, eg. "ORG=Hs" for human, etc. |
| (opt) unigeneURL | http://www.ncbi.nlm.nih.gov/UniGene/clust.cgi? ORG=Hs&CID= |
String | NCBI UNIGENE by Clone ID server. You may use an alternative UniGene server. The "ORG=Hs" should be changed to reflect the proper species, eg. "ORG=Mm" for mouse, etc. |
| (opt) locusLinkURL | http://www.ncbi.nlm.nih.gov/LocusLink/list.cgi? SITE=104&V=1&ORG=Hs&ORG=Mm&ORG=Rn&ORG=Dr&ORG=Dm&Q= |
String | NCBI LocusLink by GenBank ID server. The LocusLink server is accessed by LocusID |
| gbid2LocusLinkURL | http://www.ncbi.nlm.nih.gov/LocusLink/list.cgi?SITE=104 &V=1&ORG=Hs&ORG=Mm&ORG=Rn&ORG=Dr&ORG=Dm&Q= |
String | NCBI LocusLink by LocusID server. The LocusLink server is accessed by LocusID |
| (opt) swissProtURL | http://www.expasy.ch/cgi-bin/get-sprot-entry? | String | SwissProt by SwissProt ID |
| (opt) omimURL | http://www.ncbi.nlm.nih.gov:80/entrez/dispomim.cgi?id= | String | NCBI OMIM database by OMIM ID |
| (opt) pirURL | http://pir.georgetown.edu/cgi-bin/iproclass/iproclass?choice=entry&id= | String | PIR ProClass database by SwissProt ID |
| (opt) GeneCardURL | http://bioinfo.weizmann.ac.il/cards-bin/carddisp? | String | GeneCard DB server. You may use an alternative server. |
| (opt) histologyURL | http://mammary.nih.gov/models/ | String | E.g NIDDK MGAP histology DB server. If you have an alternative histology model server, put it here. |
| (opt) modelsURL | http://mammary.nih.gov/models/ | String | e.g. NIDDK MGAP mouse models DB server. You may use an alternative models server. |
| (opt) proxyServer | http://www.lecb.ncifcrf.gov/cgi-bin/maeProxySvr? | String | NCI/LECB proxy server to access servers outside of the Java "sandbox". If you set up MAExplorer on your local server, then] this should point to a proxy server on your system. |
| Parameter | Value | DataType | Comments |
|---|---|---|---|
| (opt) HelpMenu1 | List of hybridized samples | String | Help sub menu URL |
| (opt) HelpMenu2 | MGAP animal models | String | Help sub menu URL |
| (opt) HelpMenu3 | MGAP home page | String | Help sub menu URL |
| (opt) HelpURL1 | http://www.lecb.ncifcrf.gov/mae/maeHybridizations.html | String | Help sub menu URL |
| (opt) HelpURL2 | http://mammary.nih.gov/models/ | String | Help sub menu URL |
| (opt) HelpURL3 | http://mammary.nih.gov/ | String | Help sub menu URL |
| Parameter | Value | DataType | Comments |
|---|---|---|---|
| (opt) PluginMenuName1 | New Cluster plot | String | Plugin sub menu string |
| (opt) PluginMenuStubName1 | PlotMenu:cluster | String | name of Plugin menu stub to add menu entry |
| (opt) PluginClassFile1 | NewClusterPlot.jar | String | Name of class file |
| (opt)sPluginCallAtStartup1 | InstallInMenu | String | handling plugins at startup: "InstallInMenu", "RunOnStartup", "NoInstall" |
| (opt) PluginMenuName2 | New sample report | String | Plugin sub menu string |
| (opt) PluginMenuStubName2 | ReportMenu:sample | String | name of Plugin menu stub to add menu entry |
| (opt) PluginClassFile2 | NewSampleReport.jar | String | Name of class file |
| (opt)sPluginCallAtStartup2 | InstallInMenu | String | handling plugins at startup: "InstallInMenu", "RunOnStartup", "NoInstall" |
| (opt) PluginMenuName3 | Client-server | String | Plugin sub menu string |
| (opt) PluginMenuStubName2 | -none- | String | name of Plugin menu stub to add menu entry |
| (opt) PluginClassFile2 | ClineServerMAE.class | String | Name of class file |
| (opt)sPluginCallAtStartup2 | InstallInMenu | String | handling plugins at startup: "InstallInMenu", "RunOnStartup", "NoInstall" |
C.6 Using the Cvt2Mae 'wizard' tool to convert your array
data for use with MAExplorer
In order to use MAExplorer on your data, you must convert your data
files into the data formats described in
Appendix C and Appendix D. Although
we and others have done this by editing user's data files into the
required formats, it is a non-trivial process.
Therefore we have created a and install Cvt2Mae on your computer and use it to
convert your array data to MAExplorer data format. Figure C.6.1 shows
Cvt2Mae array data converter.
Cvt2Mae is a "Wizard" driven process designed for use by molecular biologists. It handles commercial chips such as Incyte, Affymetrix, GenePix, Scanalyze, etc. or one-of-a-kind academic chips. It asks you questions to describe your chip and your data. We call the chip description the "Array Layout". After you have created or edited an array layout, you may save it for use in future conversions. [The array layouts are kept in a subdirectory "ArrayLayout" in the directory where you installed Cvt2Mae.] Since an ArrayLayout is a file, you could mail it to a collaborator. After you have answered the questions, you then run the converter and it generates the proper set of converted data files. In the case of user defined array layouts, we denote the latter as <User-defined> where the user assigns a name to that layout as part of the description. Essentially, the array layout contains a set of "rules" for describing the user's array data so Cvt2Mae knows how to read it. At some point, we plan to add the MAGE-ML standard to Cvt2Mae as one of the array layouts so it should be able to handle a wider variety of data.
Handling grid geometries that don't fit our model
If your array geometry does not conform to one of those handled by
MAExplorer (see Section 1.1
Gene coordinate numbering on the microarray), then treat the data
as an list of spots. In Cvt2Mae Wizard panel "[2] Grid geometry data",
select the checkbox "Use # spots (BELOW), else grid-geometry (ABOVE)"
and then enter the total number of spots in the line below. This will
construct an arbitrary pseudoarray geometry to serve as a basis to
display the microarray pseudoimage (see the Algorithm for constructing
the pseudo array from a list of spots in Appendix C.6). For
example, this might be used in the case where your arrays used
meta-grids.
Figure C.6.1 The Cvt2Mae array data converter.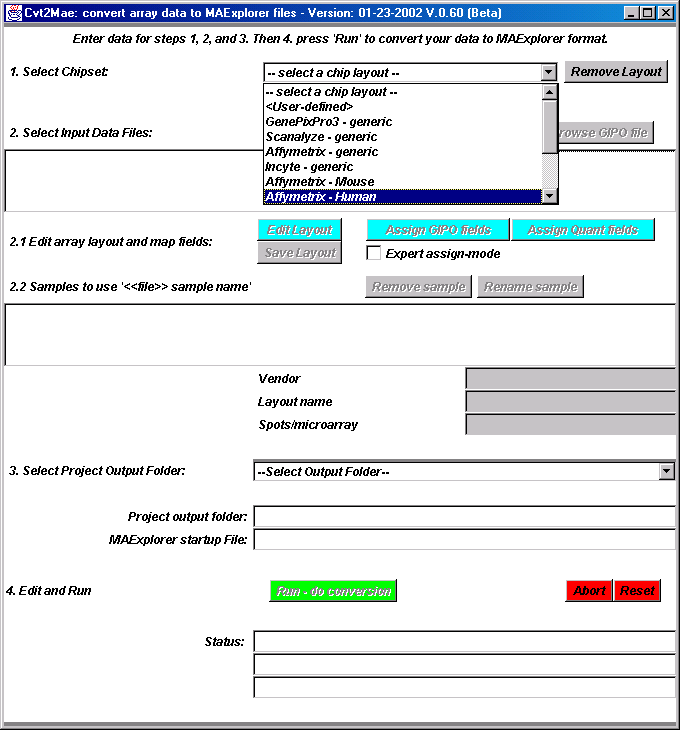
|
The details on Cvt2Mae including more description, PDF examples of conversions
for several different types of arrays, the download area, status of the converter,
etc. are available on the
Algorithm: Generation of a pseudoarray image geometry if no array geometry is specified
MAExplorer requires the data in the GIPO and Quant files be specified
by a spot position. This is indicated by the array spot geometry of
(#fields, #grids, #rows/grid, #columns/grid). The #fields is the
number of duplicated sets of grids if available - it is 1
otherwise. This 4-tuple must be specified in the Configuration file.
However, some array data does not have a spot geometry position data
available. The alternative is to generate a pseudoarray geometry. This
is possible since the pseudoarray image in MAExplorer is used simply
to indicate success of the data filter or relative differences
depending on the "Plot | Show Microarray" option. In Cvt2Mae we
generate a visually appealing pseudoarray image geometry if no array
geometry is specified with the data (e.g. Affymetrix data, etc). The
algorithm presented below will generate a geometry
(nGrids,nGridRows,nGridCols) that is compatible with the
visual use of the pseudoarray. The only assumption is the
nRowsExpected, the number of spots in the microarray (rows in
the database input file). The number of spots in the array is computed
automatically and the option to use the pseudoarray instead of the
actual array geometry is selected in the
Edit Layout Wizard for Grid Geometry.
OPT_GRID_SIZE = 1200; /* Optimal grid size for MAExplorer viewing */
ROWS_TO_COLS_ASPECT_RATIO = 3.0/4.0; /* desired rows/cols aspect aspect for a grid */
extra = 0; /* # of extra grid cols required */
/* Estimate # of grids. Assume a square aspect ratio */
if(n <= OPT_GRID_SIZE)
nGrids = 1;
else
nGrids = (n / OPT_GRID_SIZE)+1;
/* Estimate rows (r) and columns (c) from a rectangular grid
* where cols = (4/3) rows.
* Then, c = (4/3)r and r*c= area.
* Then (4/3)*r*r = area or
* r = sqrt((3/4)*area).
*/
if(nRowsExpected > 0)
while(true)
{ /* iterate to optimal size */
gridSize = n/nGrids;
nGridRows = sqrt( ROWS_TO_COLS_ASPECT_RATIO * gridSize );
nGridCols = (nGridRows / ROWS_TO_COLS_ASPECT_RATIO);
nGridCols += extra;
estTotSize = (nGrids * nGridRows * nGridCols);
if(estTotSize > nRowsExpected)
break;
else
extra++; /* keep trying until meet criteria */
} /* iterate to optimal size */
