Mozilla Public License 1.1 (MPL 1.1)
1. Definitions.
1.0.1. "Commercial Use" means distribution or otherwise making
the Covered Code available to a third party.
1.1. ''Contributor'' means each entity that creates or contributes
to the creation of Modifications.
1.2. ''Contributor Version'' means the combination of the Original
Code, prior Modifications used by a Contributor, and the Modifications
made by that particular Contributor.
1.3. ''Covered Code'' means the Original Code or Modifications
or the combination of the Original Code and Modifications, in each case
including portions thereof.
1.4. ''Electronic Distribution Mechanism'' means a mechanism
generally accepted in the software development community for the electronic
transfer of data.
1.5. ''Executable'' means Covered Code in any form other than
Source Code.
1.6. ''Initial Developer'' means the individual or entity identified
as the Initial Developer in the Source Code notice required by Exhibit
A.
1.7. ''Larger Work'' means a work which combines Covered Code
or portions thereof with code not governed by the terms of this License.
1.8. ''License'' means this document.
1.8.1. "Licensable" means having the right to grant, to the maximum
extent possible, whether at the time of the initial grant or subsequently
acquired, any and all of the rights conveyed herein.
1.9. ''Modifications'' means any addition to or deletion from
the substance or structure of either the Original Code or any previous
Modifications. When Covered Code is released as a series of files, a Modification
is:
1.10. ''Original Code'' means Source Code of computer software code
which is described in the Source Code notice required by Exhibit A
as Original Code, and which, at the time of its release under this License
is not already Covered Code governed by this License.
1.10.1. "Patent Claims" means any patent claim(s), now owned
or hereafter acquired, including without limitation, method, process,
and apparatus claims, in any patent Licensable by grantor.
1.11. ''Source Code'' means the preferred form of the Covered
Code for making modifications to it, including all modules it contains,
plus any associated interface definition files, scripts used to control
compilation and installation of an Executable, or source code differential
comparisons against either the Original Code or another well known, available
Covered Code of the Contributor's choice. The Source Code can be in a compressed
or archival form, provided the appropriate decompression or de-archiving
software is widely available for no charge.
1.12. "You'' (or "Your") means an individual or a legal
entity exercising rights under, and complying with all of the terms of,
this License or a future version of this License issued under Section 6.1.
For legal entities, "You'' includes any entity which controls, is controlled
by, or is under common control with You. For purposes of this definition,
"control'' means (a) the power, direct or indirect, to cause the direction
or management of such entity, whether by contract or otherwise, or (b)
ownership of more than fifty percent (50%) of the outstanding shares or
beneficial ownership of such entity.
2. Source Code License.
2.1. The Initial Developer Grant.
The Initial Developer hereby grants You a world-wide, royalty-free,
non-exclusive license, subject to third party intellectual property claims:
(a) under intellectual property rights (other than
patent or trademark) Licensable by Initial Developer to use, reproduce,
modify, display, perform, sublicense and distribute the Original Code (or
portions thereof) with or without Modifications, and/or as part of a Larger
Work; and
(b) under Patents Claims infringed by the making, using or selling
of Original Code, to make, have made, use, practice, sell, and offer for
sale, and/or otherwise dispose of the Original Code (or portions thereof).
(c) the licenses granted in this Section 2.1(a) and (b) are effective
on the date Initial Developer first distributes Original Code under the
terms of this License.
(d) Notwithstanding Section 2.1(b) above, no patent license is
granted: 1) for code that You delete from the Original Code; 2) separate
from the Original Code; or 3) for infringements caused by: i) the
modification of the Original Code or ii) the combination of the Original
Code with other software or devices.
2.2. Contributor Grant.
Subject to third party intellectual property claims, each Contributor
hereby grants You a world-wide, royalty-free, non-exclusive license
(a) under intellectual property rights (other than
patent or trademark) Licensable by Contributor, to use, reproduce, modify,
display, perform, sublicense and distribute the Modifications created by
such Contributor (or portions thereof) either on an unmodified basis, with
other Modifications, as Covered Code and/or as part of a Larger Work; and
(b) under Patent Claims infringed by the making, using, or selling
of Modifications made by that Contributor either alone and/or in
combination with its Contributor Version (or portions of such combination),
to make, use, sell, offer for sale, have made, and/or otherwise dispose
of: 1) Modifications made by that Contributor (or portions thereof); and
2) the combination of Modifications made by that Contributor with
its Contributor Version (or portions of such combination).
(c) the licenses granted in Sections 2.2(a) and 2.2(b) are effective
on the date Contributor first makes Commercial Use of the Covered Code.
(d) Notwithstanding Section 2.2(b) above, no
patent license is granted: 1) for any code that Contributor has deleted
from the Contributor Version; 2) separate from the Contributor Version;
3) for infringements caused by: i) third party modifications of Contributor
Version or ii) the combination of Modifications made by that Contributor
with other software (except as part of the Contributor Version) or
other devices; or 4) under Patent Claims infringed by Covered Code in the
absence of Modifications made by that Contributor.
3. Distribution Obligations.
3.1. Application of License.
The Modifications which You create or to which You contribute are governed
by the terms of this License, including without limitation Section 2.2.
The Source Code version of Covered Code may be distributed only under the
terms of this License or a future version of this License released under
Section 6.1, and You must include a copy of this License with every
copy of the Source Code You distribute. You may not offer or impose any
terms on any Source Code version that alters or restricts the applicable
version of this License or the recipients' rights hereunder. However, You
may include an additional document offering the additional rights described
in Section 3.5.
3.2. Availability of Source Code.
Any Modification which You create or to which You contribute must be
made available in Source Code form under the terms of this License either
on the same media as an Executable version or via an accepted Electronic
Distribution Mechanism to anyone to whom you made an Executable version
available; and if made available via Electronic Distribution Mechanism,
must remain available for at least twelve (12) months after the date it
initially became available, or at least six (6) months after a subsequent
version of that particular Modification has been made available to such
recipients. You are responsible for ensuring that the Source Code version
remains available even if the Electronic Distribution Mechanism is maintained
by a third party.
3.3. Description of Modifications.
You must cause all Covered Code to which You contribute to contain
a file documenting the changes You made to create that Covered Code and
the date of any change. You must include a prominent statement that the
Modification is derived, directly or indirectly, from Original Code provided
by the Initial Developer and including the name of the Initial Developer
in (a) the Source Code, and (b) in any notice in an Executable version
or related documentation in which You describe the origin or ownership
of the Covered Code.
3.4. Intellectual Property Matters
(a) Third Party Claims.
If Contributor has knowledge that a license under a third party's intellectual
property rights is required to exercise the rights granted by such Contributor
under Sections 2.1 or 2.2, Contributor must include a text file with the
Source Code distribution titled "LEGAL'' which describes the claim and
the party making the claim in sufficient detail that a recipient will know
whom to contact. If Contributor obtains such knowledge after the Modification
is made available as described in Section 3.2, Contributor shall promptly
modify the LEGAL file in all copies Contributor makes available thereafter
and shall take other steps (such as notifying appropriate mailing lists
or newsgroups) reasonably calculated to inform those who received the Covered
Code that new knowledge has been obtained.
(b) Contributor APIs.
If Contributor's Modifications include an application programming interface
and Contributor has knowledge of patent licenses which are reasonably necessary
to implement that API, Contributor must also include this information in
the LEGAL file.
(c)
Representations.
Contributor represents that, except as disclosed pursuant to Section
3.4(a) above, Contributor believes that Contributor's Modifications are
Contributor's original creation(s) and/or Contributor has sufficient rights
to grant the rights conveyed by this License.
3.5. Required Notices.
You must duplicate the notice in Exhibit A in each file of the
Source Code. If it is not possible to put such notice in a particular
Source Code file due to its structure, then You must include such notice
in a location (such as a relevant directory) where a user would be likely
to look for such a notice. If You created one or more Modification(s)
You may add your name as a Contributor to the notice described in Exhibit
A. You must also duplicate this License in any documentation
for the Source Code where You describe recipients' rights or ownership
rights relating to Covered Code. You may choose to offer, and to
charge a fee for, warranty, support, indemnity or liability obligations
to one or more recipients of Covered Code. However, You may do so only
on Your own behalf, and not on behalf of the Initial Developer or any Contributor.
You must make it absolutely clear than any such warranty, support, indemnity
or liability obligation is offered by You alone, and You hereby agree to
indemnify the Initial Developer and every Contributor for any liability
incurred by the Initial Developer or such Contributor as a result of warranty,
support, indemnity or liability terms You offer.
3.6. Distribution of Executable Versions.
You may distribute Covered Code in Executable form only if the requirements
of Section 3.1-3.5 have been met for that Covered Code, and if You
include a notice stating that the Source Code version of the Covered Code
is available under the terms of this License, including a description of
how and where You have fulfilled the obligations of Section 3.2.
The notice must be conspicuously included in any notice in an Executable
version, related documentation or collateral in which You describe recipients'
rights relating to the Covered Code. You may distribute the Executable
version of Covered Code or ownership rights under a license of Your choice,
which may contain terms different from this License, provided that You
are in compliance with the terms of this License and that the license for
the Executable version does not attempt to limit or alter the recipient's
rights in the Source Code version from the rights set forth in this License.
If You distribute the Executable version under a different license You
must make it absolutely clear that any terms which differ from this License
are offered by You alone, not by the Initial Developer or any Contributor.
You hereby agree to indemnify the Initial Developer and every Contributor
for any liability incurred by the Initial Developer or such Contributor
as a result of any such terms You offer.
3.7. Larger Works.
You may create a Larger Work by combining Covered Code with other code
not governed by the terms of this License and distribute the Larger Work
as a single product. In such a case, You must make sure the requirements
of this License are fulfilled for the Covered Code.
4. Inability to Comply Due to Statute or Regulation.
If it is impossible for You to comply with any of the terms of this
License with respect to some or all of the Covered Code due to statute,
judicial order, or regulation then You must: (a) comply with the terms
of this License to the maximum extent possible; and (b) describe the limitations
and the code they affect. Such description must be included in the LEGAL
file described in Section 3.4 and must be included with all distributions
of the Source Code. Except to the extent prohibited by statute or regulation,
such description must be sufficiently detailed for a recipient of ordinary
skill to be able to understand it.
5. Application of this License.
This License applies to code to which the Initial Developer has attached
the notice in Exhibit A and to related Covered Code.
6. Versions of the License.
6.1. New Versions.
Netscape Communications Corporation (''Netscape'') may publish revised
and/or new versions of the License from time to time. Each version will
be given a distinguishing version number.
6.2. Effect of New Versions.
Once Covered Code has been published under a particular version of
the License, You may always continue to use it under the terms of that
version. You may also choose to use such Covered Code under the terms of
any subsequent version of the License published by Netscape. No one other
than Netscape has the right to modify the terms applicable to Covered Code
created under this License.
6.3. Derivative Works.
If You create or use a modified version of this License (which you
may only do in order to apply it to code which is not already Covered Code
governed by this License), You must (a) rename Your license so that the
phrases ''Mozilla'', ''MOZILLAPL'', ''MOZPL'', ''Netscape'', "MPL", ''NPL''
or any confusingly similar phrase do not appear in your license (except
to note that your license differs from this License) and (b) otherwise
make it clear that Your version of the license contains terms which differ
from the Mozilla Public License and Netscape Public License. (Filling in
the name of the Initial Developer, Original Code or Contributor in the
notice described in Exhibit A shall not of themselves be deemed
to be modifications of this License.)
7. DISCLAIMER OF WARRANTY.
COVERED CODE IS PROVIDED UNDER THIS LICENSE ON AN "AS IS'' BASIS, WITHOUT
WARRANTY OF ANY KIND, EITHER EXPRESSED OR IMPLIED, INCLUDING, WITHOUT LIMITATION,
WARRANTIES THAT THE COVERED CODE IS FREE OF DEFECTS, MERCHANTABLE, FIT
FOR A PARTICULAR PURPOSE OR NON-INFRINGING. THE ENTIRE RISK AS TO THE QUALITY
AND PERFORMANCE OF THE COVERED CODE IS WITH YOU. SHOULD ANY COVERED CODE
PROVE DEFECTIVE IN ANY RESPECT, YOU (NOT THE INITIAL DEVELOPER OR ANY OTHER
CONTRIBUTOR) ASSUME THE COST OF ANY NECESSARY SERVICING, REPAIR OR CORRECTION.
THIS DISCLAIMER OF WARRANTY CONSTITUTES AN ESSENTIAL PART OF THIS LICENSE.
NO USE OF ANY COVERED CODE IS AUTHORIZED HEREUNDER EXCEPT UNDER THIS DISCLAIMER.
8. TERMINATION.
8.1. This License and the rights granted hereunder will
terminate automatically if You fail to comply with terms herein and fail
to cure such breach within 30 days of becoming aware of the breach. All
sublicenses to the Covered Code which are properly granted shall survive
any termination of this License. Provisions which, by their nature, must
remain in effect beyond the termination of this License shall survive.
8.2. If You initiate litigation by asserting a patent infringement
claim (excluding declatory judgment actions) against Initial Developer
or a Contributor (the Initial Developer or Contributor against whom You
file such action is referred to as "Participant") alleging that:
(a) such Participant's Contributor Version directly or
indirectly infringes any patent, then any and all rights granted by such
Participant to You under Sections 2.1 and/or 2.2 of this License shall,
upon 60 days notice from Participant terminate prospectively, unless if
within 60 days after receipt of notice You either: (i) agree in writing
to pay Participant a mutually agreeable reasonable royalty for Your past
and future use of Modifications made by such Participant, or (ii) withdraw
Your litigation claim with respect to the Contributor Version against such
Participant. If within 60 days of notice, a reasonable royalty and
payment arrangement are not mutually agreed upon in writing by the parties
or the litigation claim is not withdrawn, the rights granted by Participant
to You under Sections 2.1 and/or 2.2 automatically terminate at the expiration
of the 60 day notice period specified above.
(b) any software, hardware, or device, other than such
Participant's Contributor Version, directly or indirectly infringes any
patent, then any rights granted to You by such Participant under Sections
2.1(b) and 2.2(b) are revoked effective as of the date You first made,
used, sold, distributed, or had made, Modifications made by that Participant.
8.3. If You assert a patent infringement claim against
Participant alleging that such Participant's Contributor Version directly
or indirectly infringes any patent where such claim is resolved (such as
by license or settlement) prior to the initiation of patent infringement
litigation, then the reasonable value of the licenses granted by such Participant
under Sections 2.1 or 2.2 shall be taken into account in determining the
amount or value of any payment or license.
8.4. In the event of termination under Sections 8.1 or
8.2 above, all end user license agreements (excluding distributors
and resellers) which have been validly granted by You or any distributor
hereunder prior to termination shall survive termination.
9. LIMITATION OF LIABILITY.
UNDER NO CIRCUMSTANCES AND UNDER NO LEGAL THEORY, WHETHER TORT (INCLUDING
NEGLIGENCE), CONTRACT, OR OTHERWISE, SHALL YOU, THE INITIAL DEVELOPER,
ANY OTHER CONTRIBUTOR, OR ANY DISTRIBUTOR OF COVERED CODE, OR ANY SUPPLIER
OF ANY OF SUCH PARTIES, BE LIABLE TO ANY PERSON FOR ANY INDIRECT, SPECIAL,
INCIDENTAL, OR CONSEQUENTIAL DAMAGES OF ANY CHARACTER INCLUDING, WITHOUT
LIMITATION, DAMAGES FOR LOSS OF GOODWILL, WORK STOPPAGE, COMPUTER FAILURE
OR MALFUNCTION, OR ANY AND ALL OTHER COMMERCIAL DAMAGES OR LOSSES, EVEN
IF SUCH PARTY SHALL HAVE BEEN INFORMED OF THE POSSIBILITY OF SUCH DAMAGES.
THIS LIMITATION OF LIABILITY SHALL NOT APPLY TO LIABILITY FOR DEATH OR
PERSONAL INJURY RESULTING FROM SUCH PARTY'S NEGLIGENCE TO THE EXTENT APPLICABLE
LAW PROHIBITS SUCH LIMITATION. SOME JURISDICTIONS DO NOT ALLOW THE EXCLUSION
OR LIMITATION OF INCIDENTAL OR CONSEQUENTIAL DAMAGES, SO THIS EXCLUSION
AND LIMITATION MAY NOT APPLY TO YOU.
10. U.S. GOVERNMENT END USERS.
The Covered Code is a ''commercial item,'' as that term is defined
in 48 C.F.R. 2.101 (Oct. 1995), consisting of ''commercial computer software''
and ''commercial computer software documentation,'' as such terms are used
in 48 C.F.R. 12.212 (Sept. 1995). Consistent with 48 C.F.R. 12.212 and
48 C.F.R. 227.7202-1 through 227.7202-4 (June 1995), all U.S. Government
End Users acquire Covered Code with only those rights set forth herein.
11. MISCELLANEOUS.
This License represents the complete agreement concerning subject matter
hereof. If any provision of this License is held to be unenforceable, such
provision shall be reformed only to the extent necessary to make it enforceable.
This License shall be governed by California law provisions (except to
the extent applicable law, if any, provides otherwise), excluding its conflict-of-law
provisions. With respect to disputes in which at least one party is a citizen
of, or an entity chartered or registered to do business in the United States
of America, any litigation relating to this License shall be subject to
the jurisdiction of the Federal Courts of the Northern District of California,
with venue lying in Santa Clara County, California, with the losing party
responsible for costs, including without limitation, court costs and reasonable
attorneys' fees and expenses. The application of the United Nations Convention
on Contracts for the International Sale of Goods is expressly excluded.
Any law or regulation which provides that the language of a contract shall
be construed against the drafter shall not apply to this License.
12. RESPONSIBILITY FOR CLAIMS.
As between Initial Developer and the Contributors, each party is responsible
for claims and damages arising, directly or indirectly, out of its utilization
of rights under this License and You agree to work with Initial Developer
and Contributors to distribute such responsibility on an equitable basis.
Nothing herein is intended or shall be deemed to constitute any admission
of liability.
13. MULTIPLE-LICENSED CODE.
Initial Developer may designate portions of the Covered Code as �Multiple-Licensed�.
�Multiple-Licensed� means that the Initial Developer permits you to utilize
portions of the Covered Code under Your choice of the NPL or the alternative
licenses, if any, specified by the Initial Developer in the file described
in Exhibit A.
EXHIBIT A -Mozilla Public License.
``The contents of this file are subject to the Mozilla Public License
Version 1.1 (the "License"); you may not use this file except in compliance
with the License. You may obtain a copy of the License at
http://www.mozilla.org/MPL/
Software distributed under the License is distributed on an "AS IS"
basis, WITHOUT WARRANTY OF
ANY KIND, either express or implied. See the License for the specific
language governing rights and
limitations under the License.
The Original Code is ______________________________________.
The Initial Developer of the Original Code is ________________________.
Portions created by
______________________ are Copyright (C) ______ _______________________.
All Rights
Reserved.
Contributor(s): ______________________________________.
Alternatively, the contents of this file may be used under the terms
of the _____ license (the �[___] License�), in which case the provisions
of [______] License are applicable instead of those above.
If you wish to allow use of your version of this file only under the terms
of the [____] License and not to allow others to use your version of this
file under the MPL, indicate your decision by deleting the provisions
above and replace them with the notice and other provisions required
by the [___] License. If you do not delete the provisions above,
a recipient may use your version of this file under either the MPL or the
[___] License."
[NOTE: The text of this Exhibit A may differ slightly from the text
of the notices in the Source Code files of the Original Code. You should
use the text of this Exhibit A rather than the text found in the Original
Code Source Code for Your Modifications.]
|
The U.S. Government LEGAL
notice accompanies the MPL 1.1 document.
opensource.org home page
|



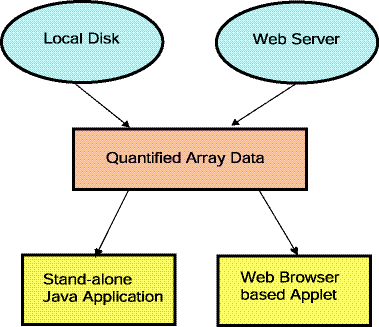
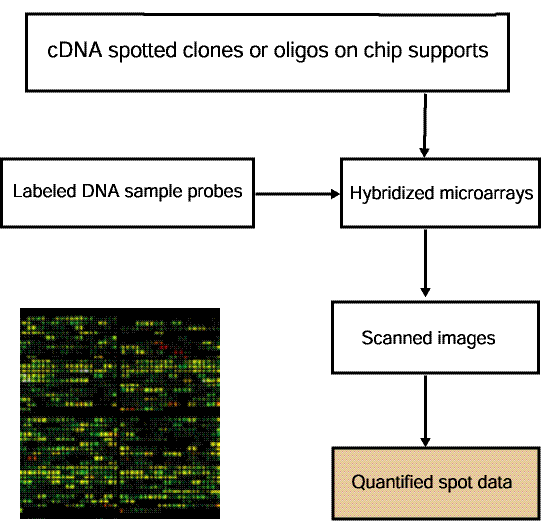
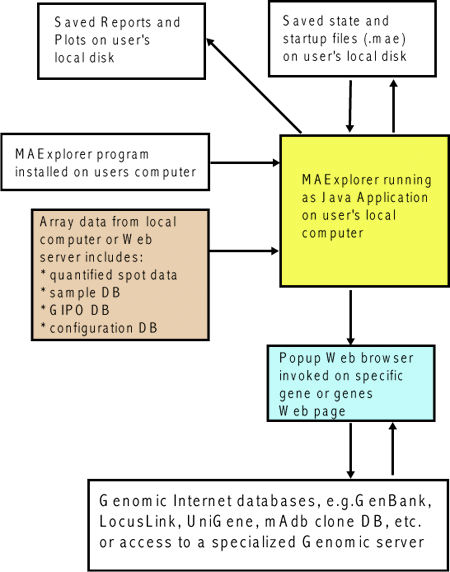
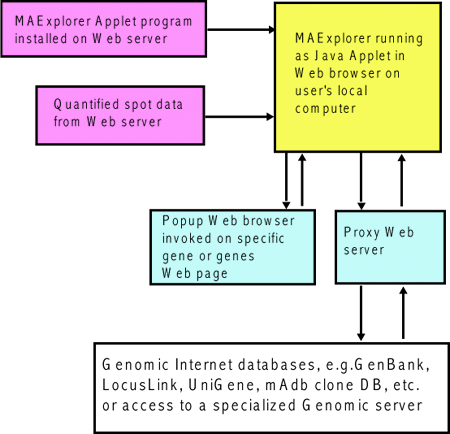



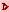 '. Selections prefaced with a '
'. Selections prefaced with a '
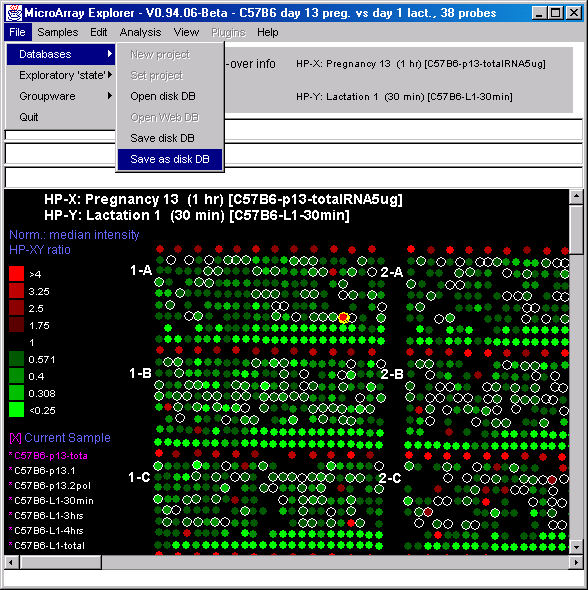

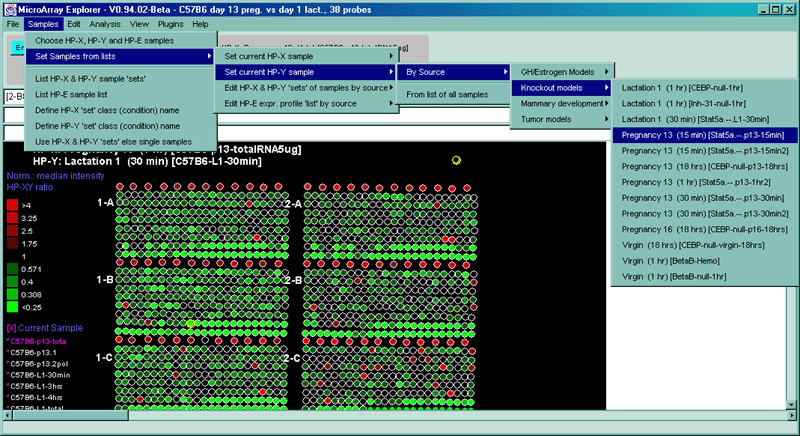
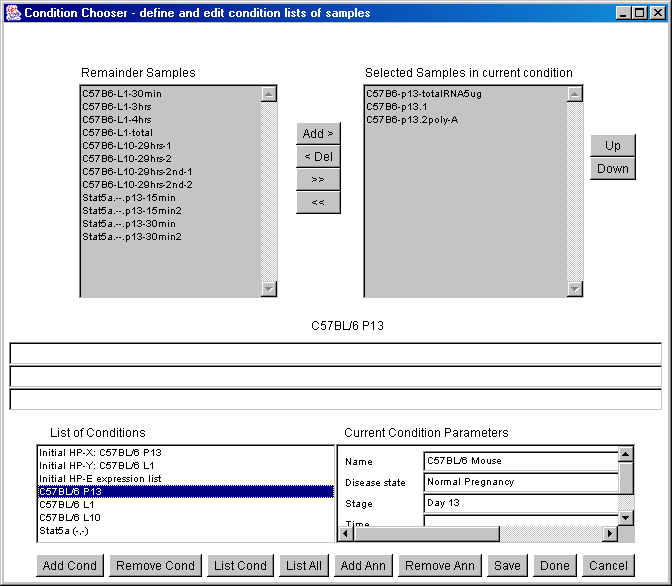
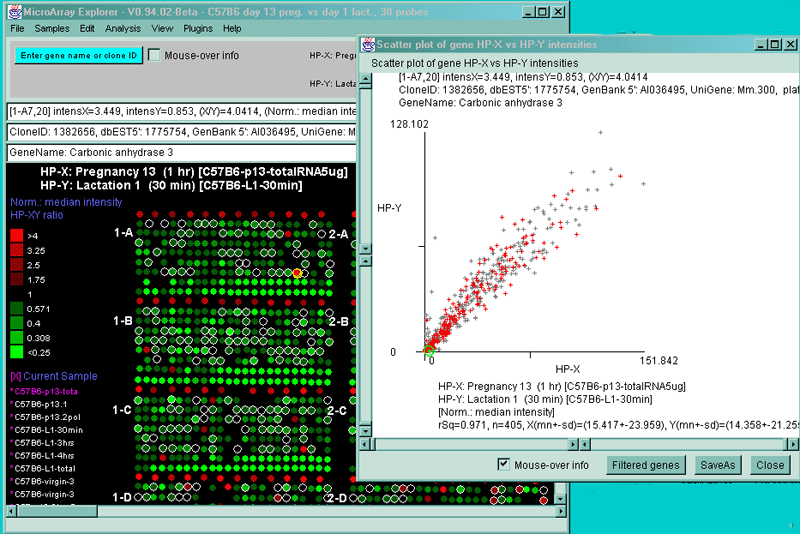
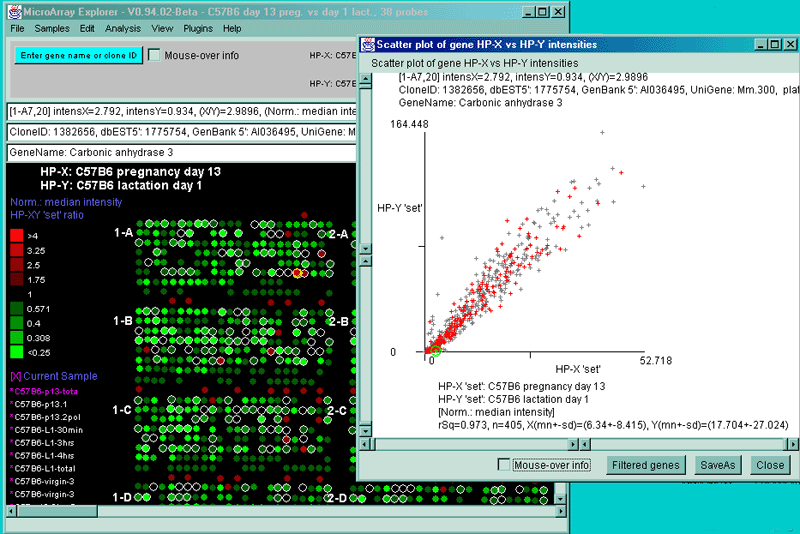
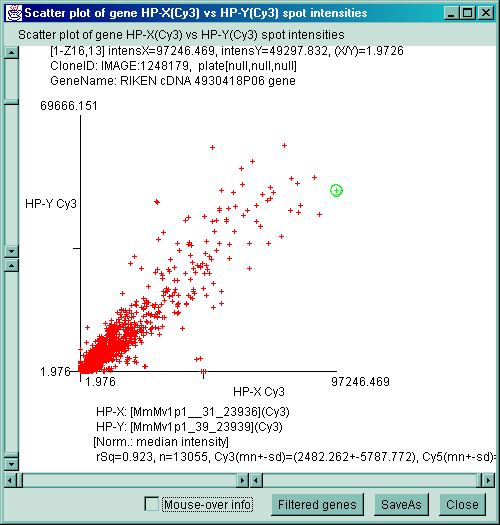
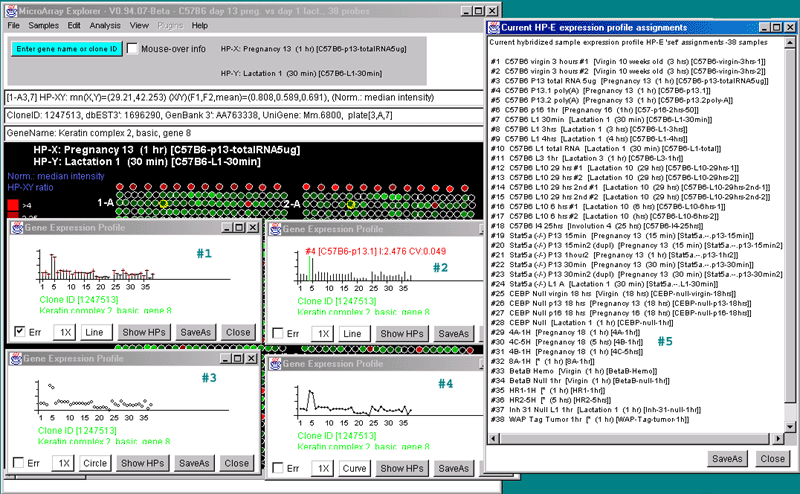
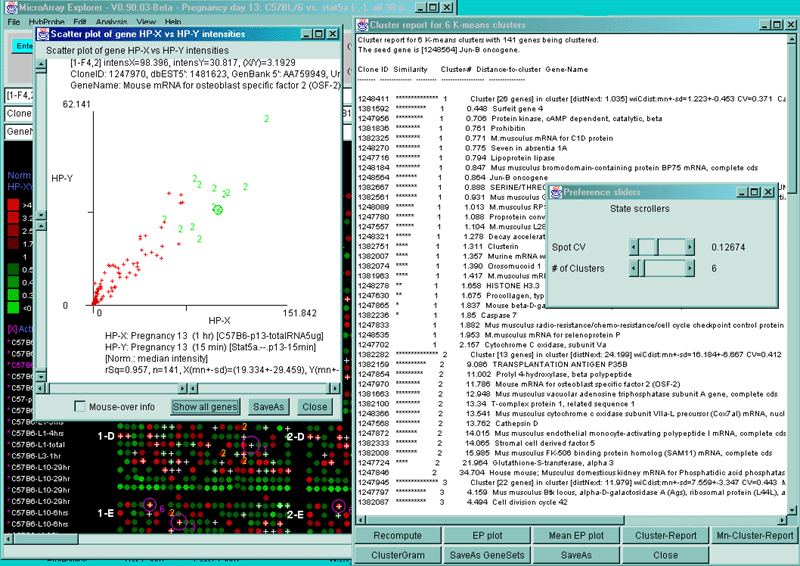
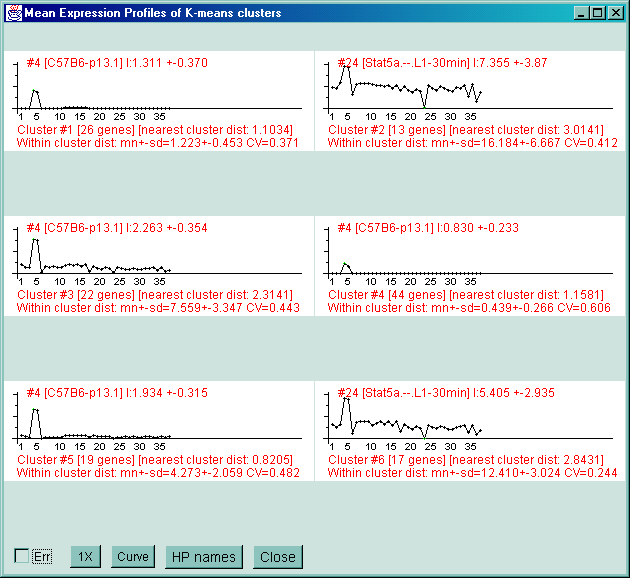
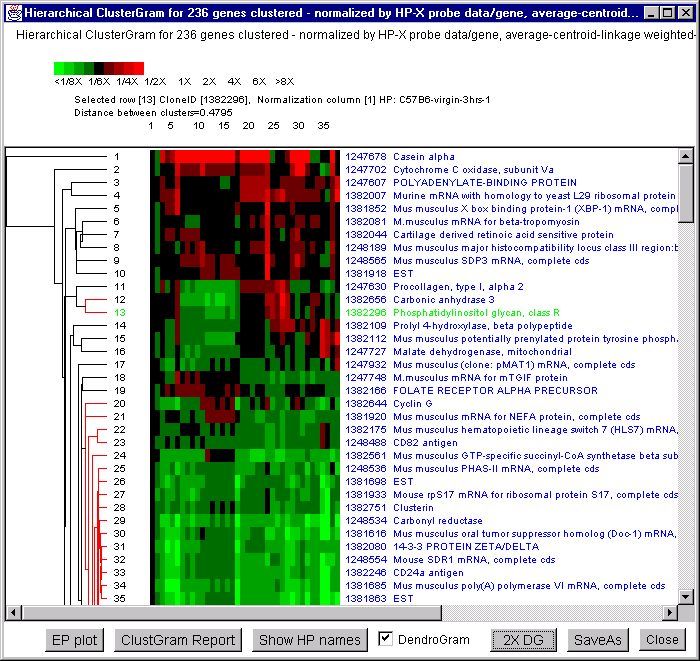
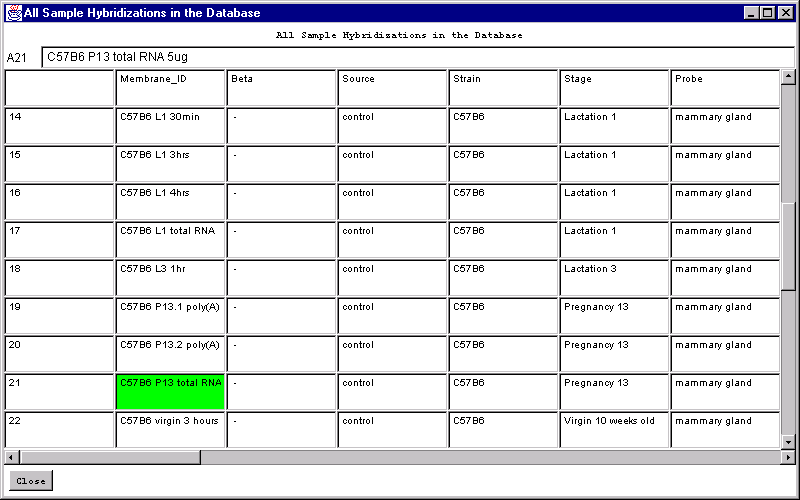
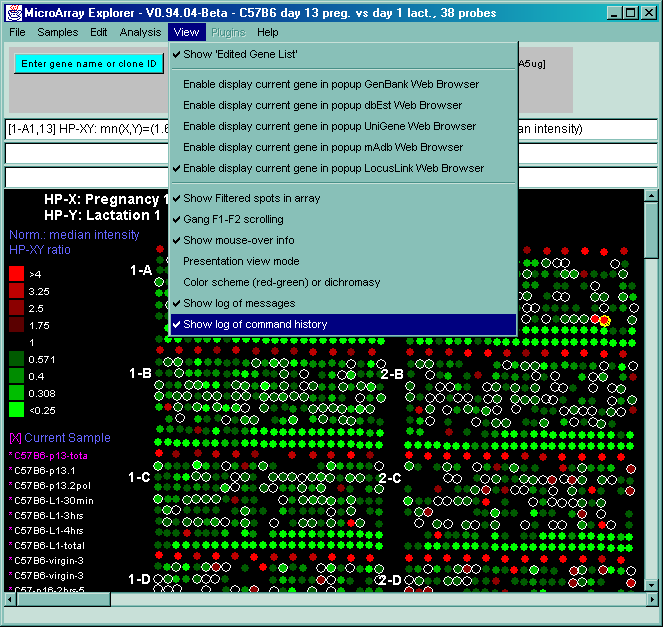
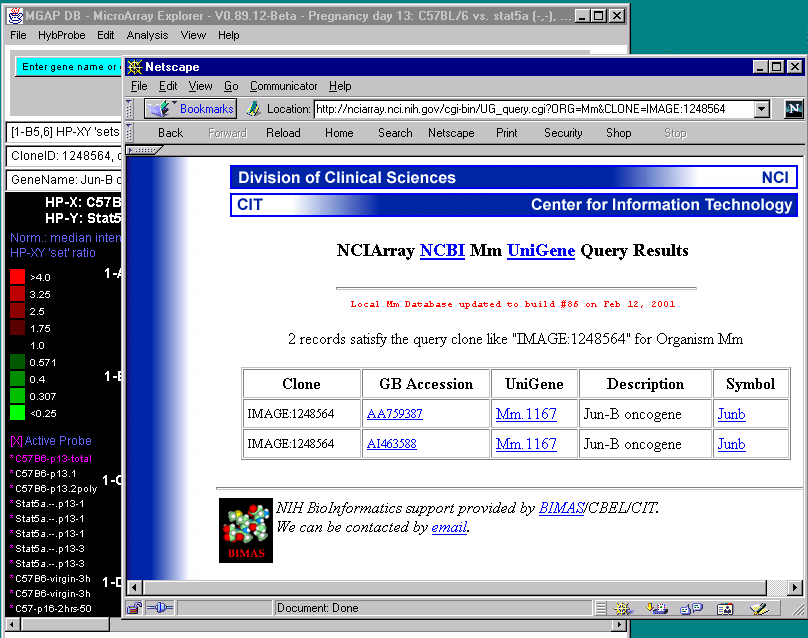
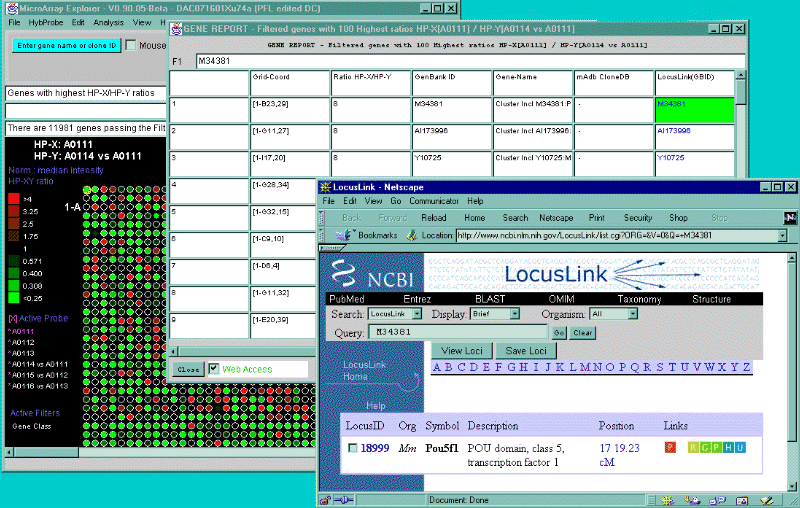
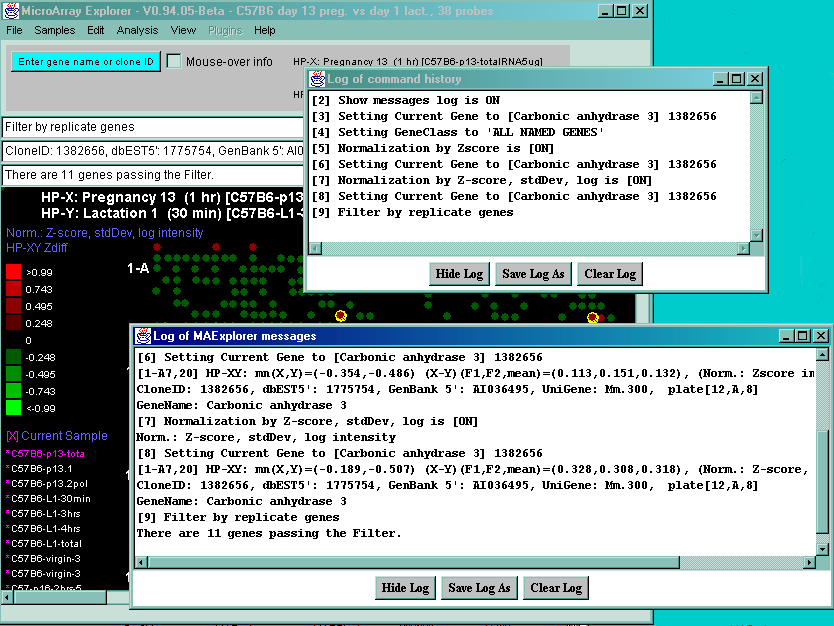
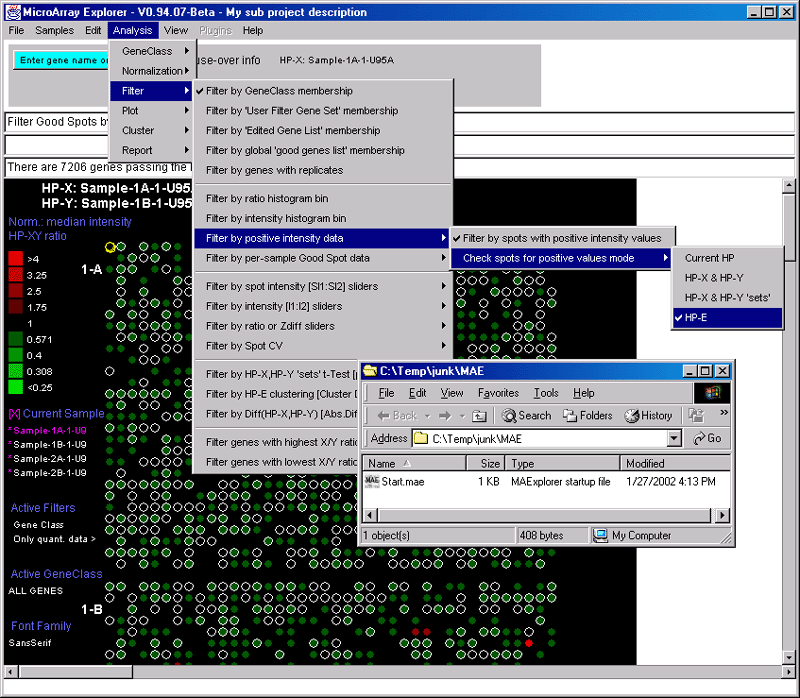
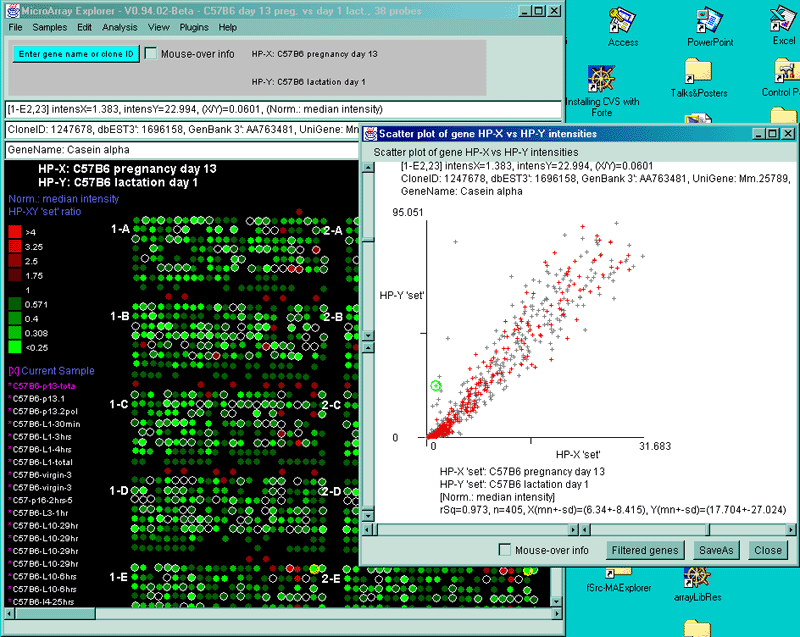
![Switching current X or Y sample by clicking on desired sample
in list at left edge of pseudoarray image. Switch between
X and Y by toggling '[X]' and '[Y]' of the Current Sample box.](GifsDocH/fig-HybProbeSetFromImageLarge.gif)









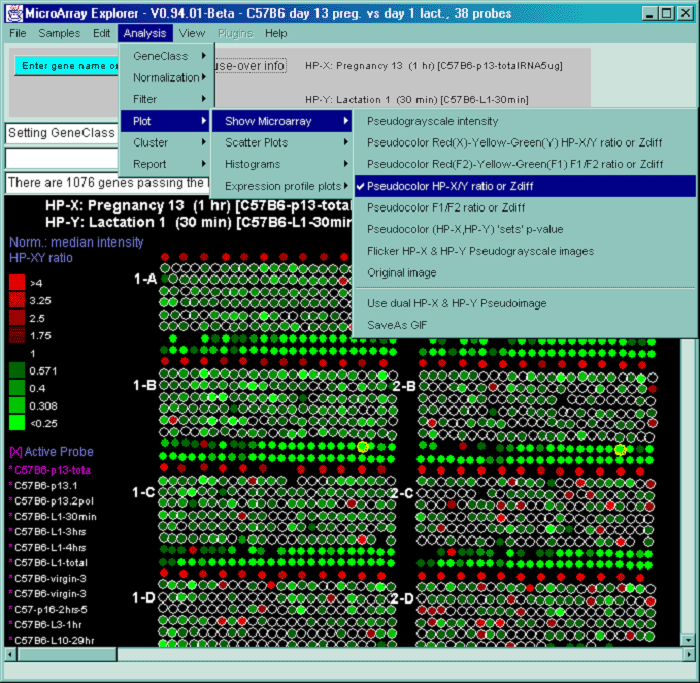
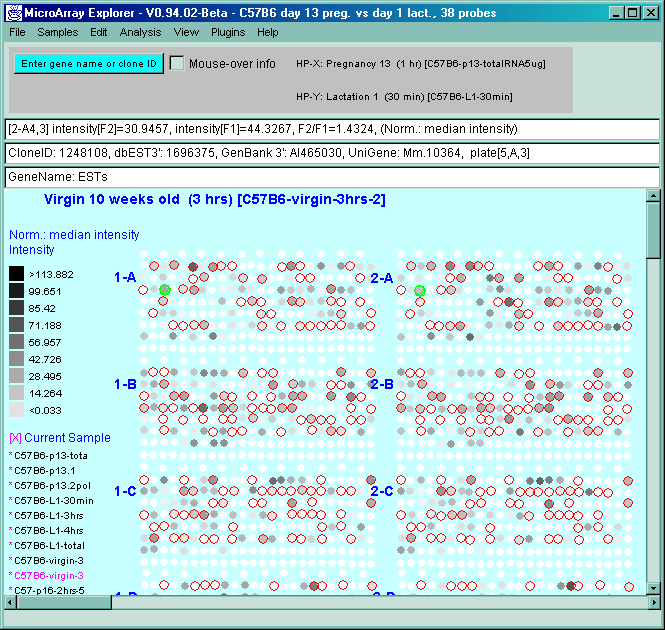

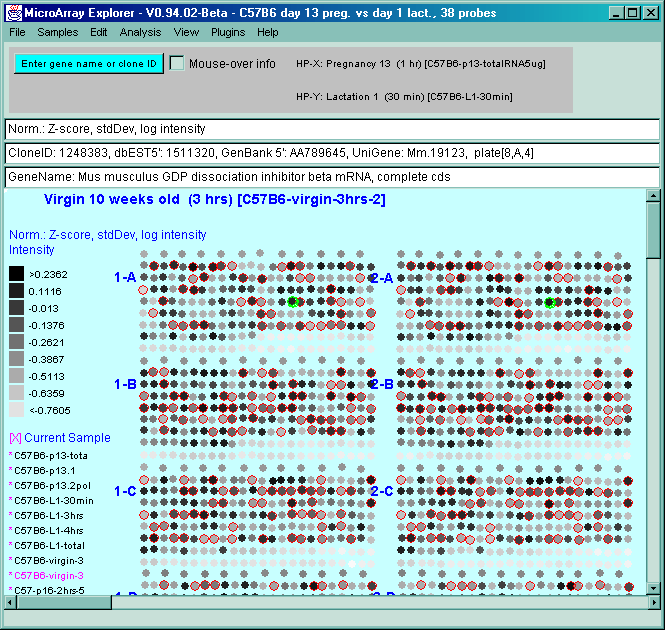
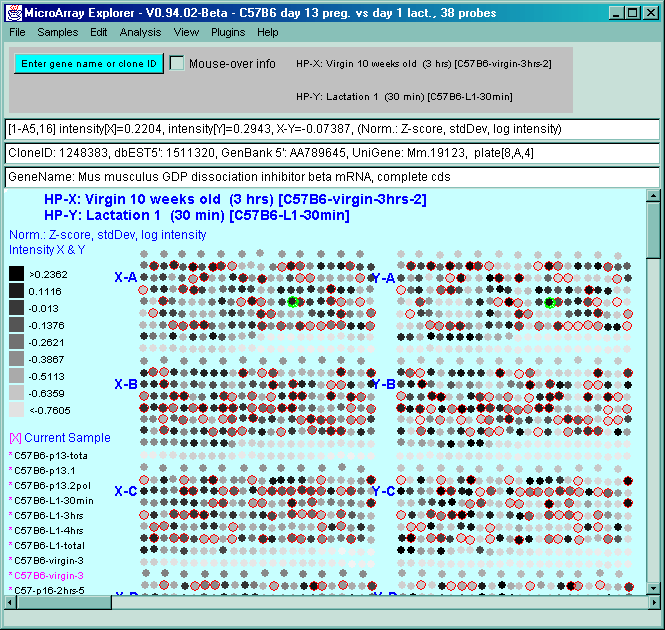
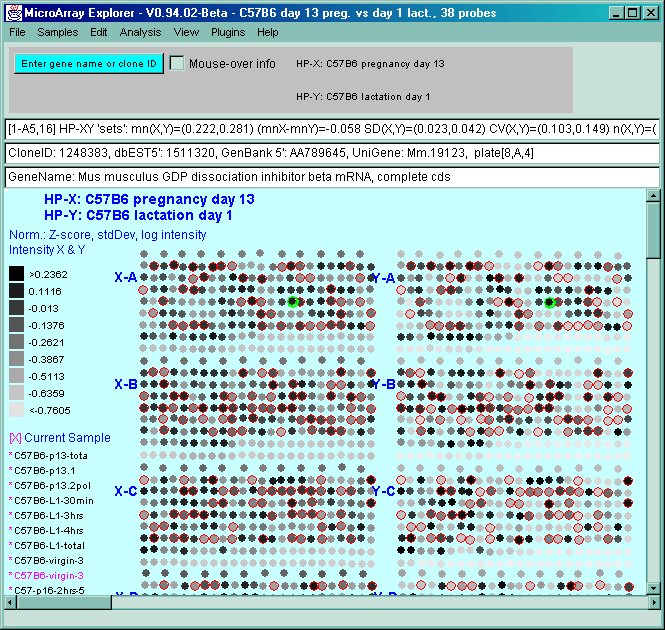

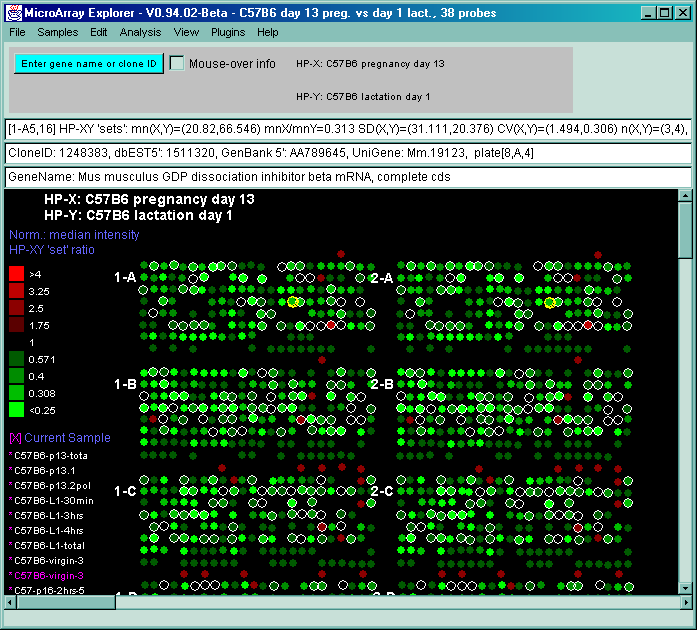
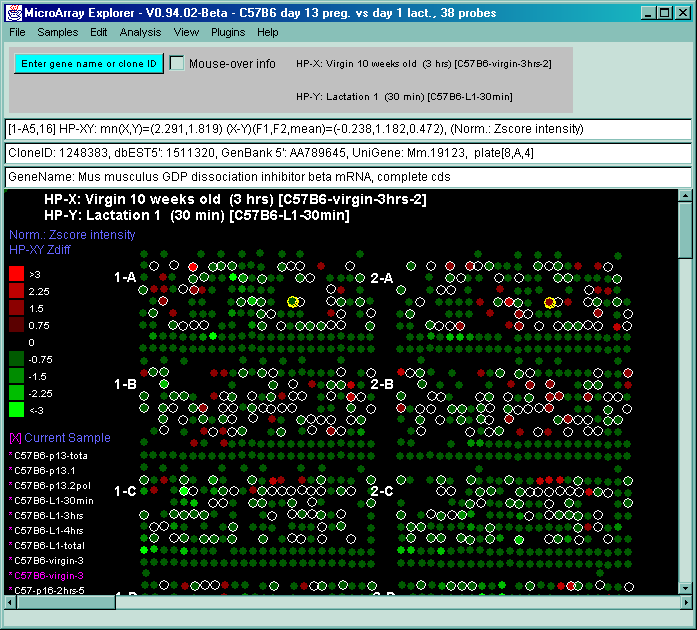
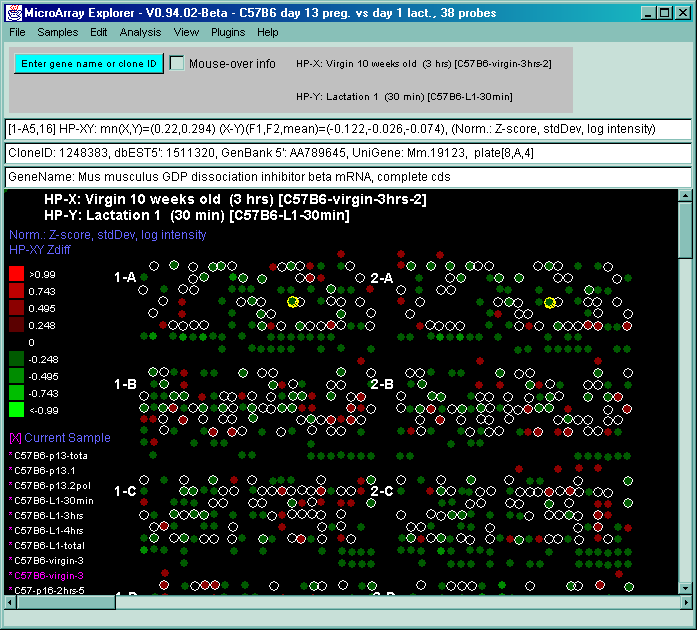










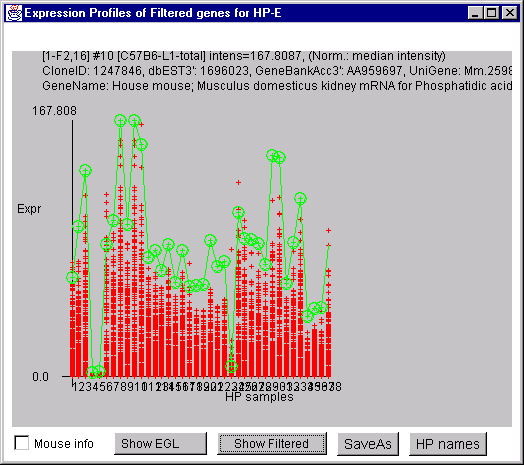
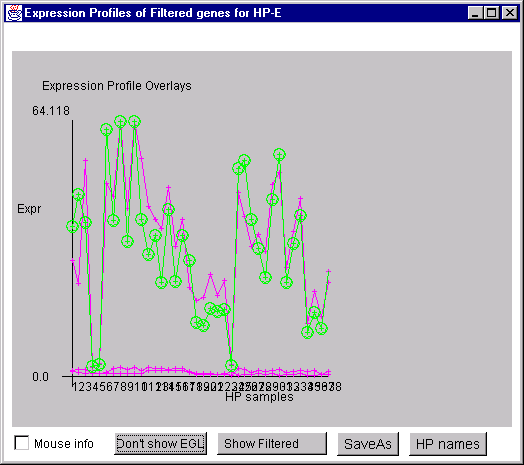

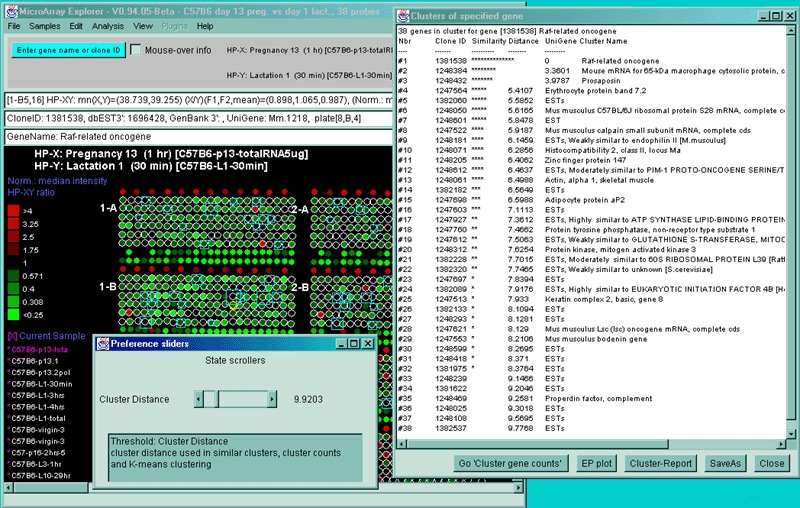

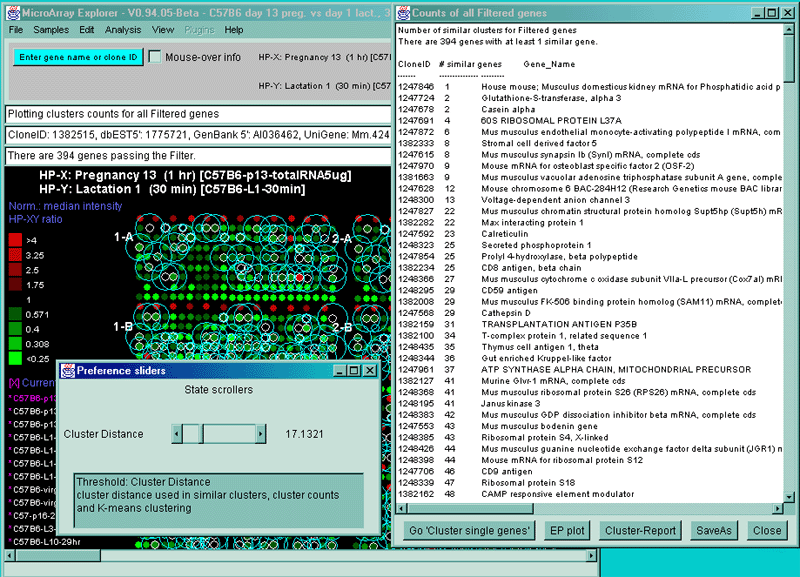






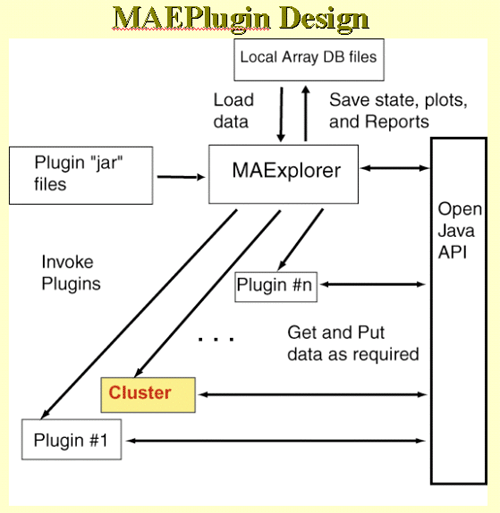
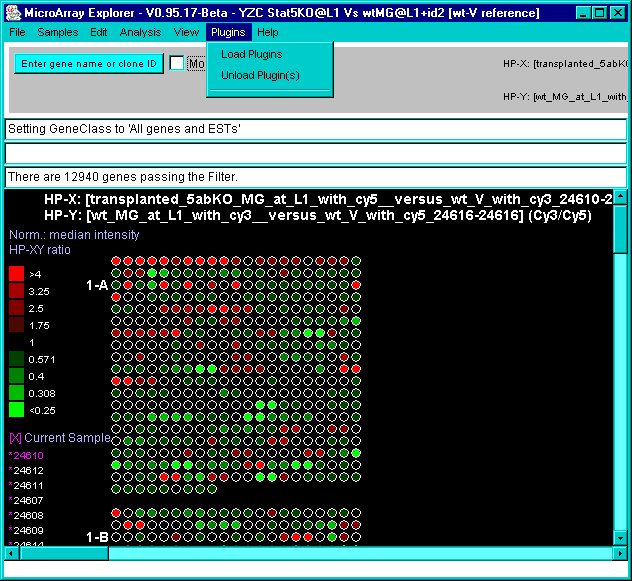


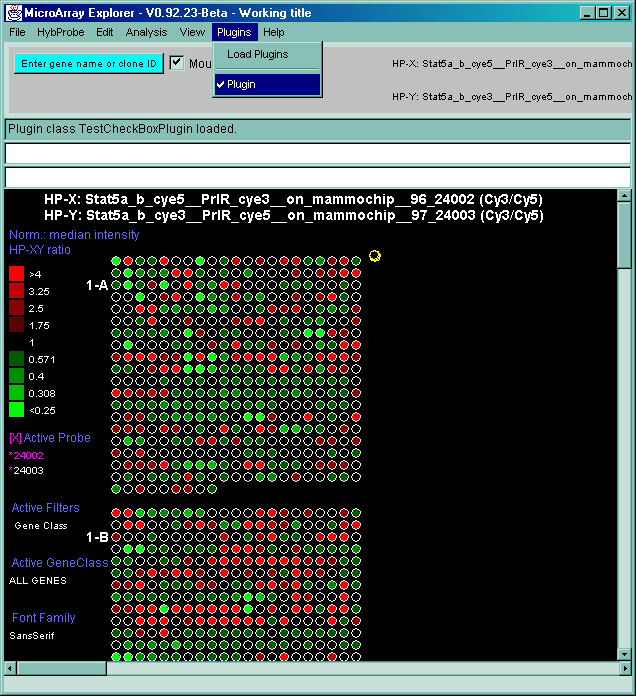

 4.1 Known Bugs in MAExplorer
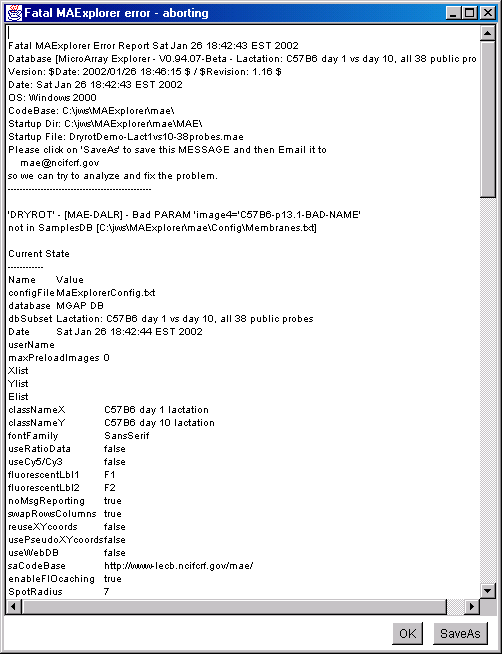
4.1 Known Bugs in MAExplorer




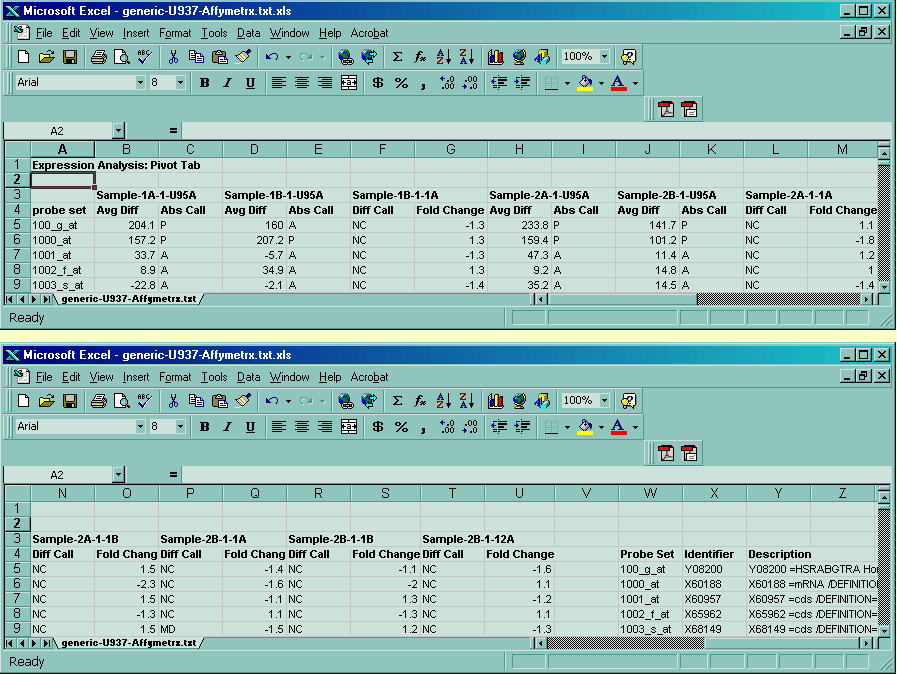

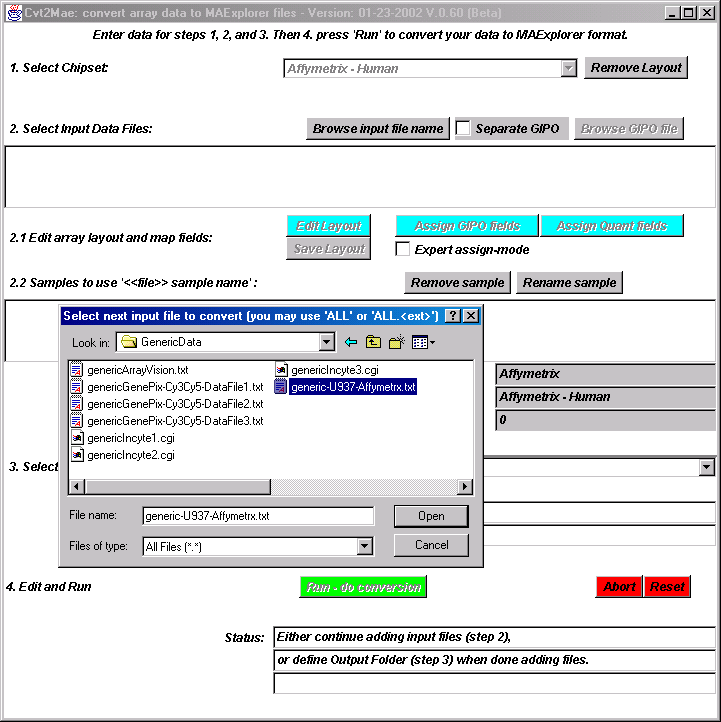
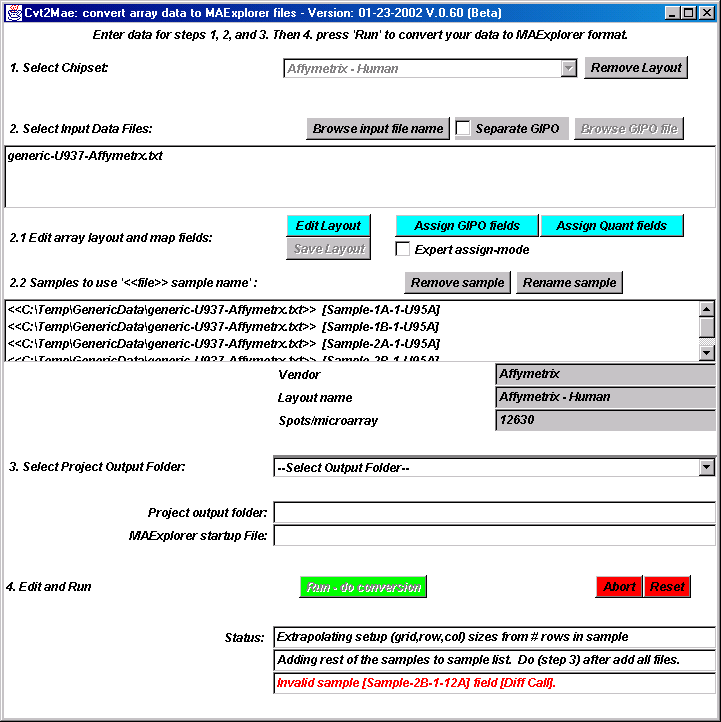






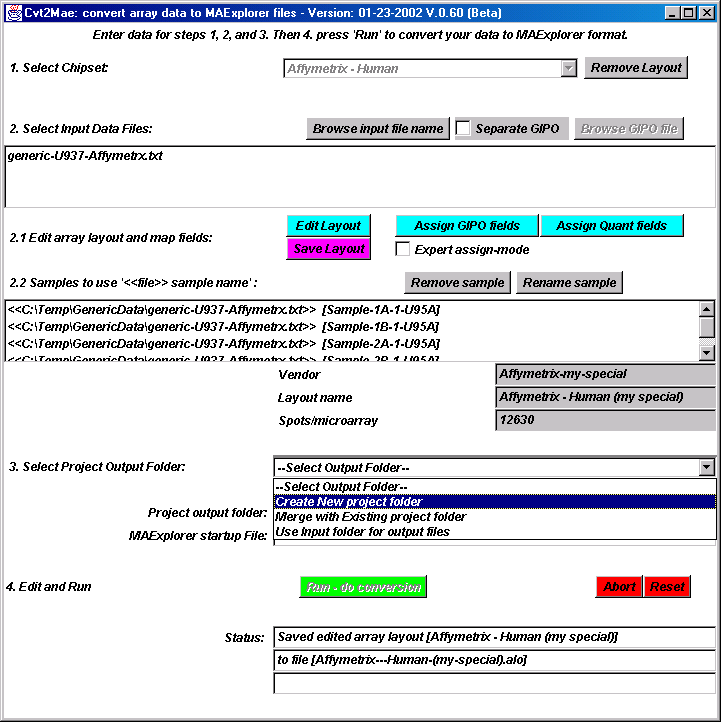
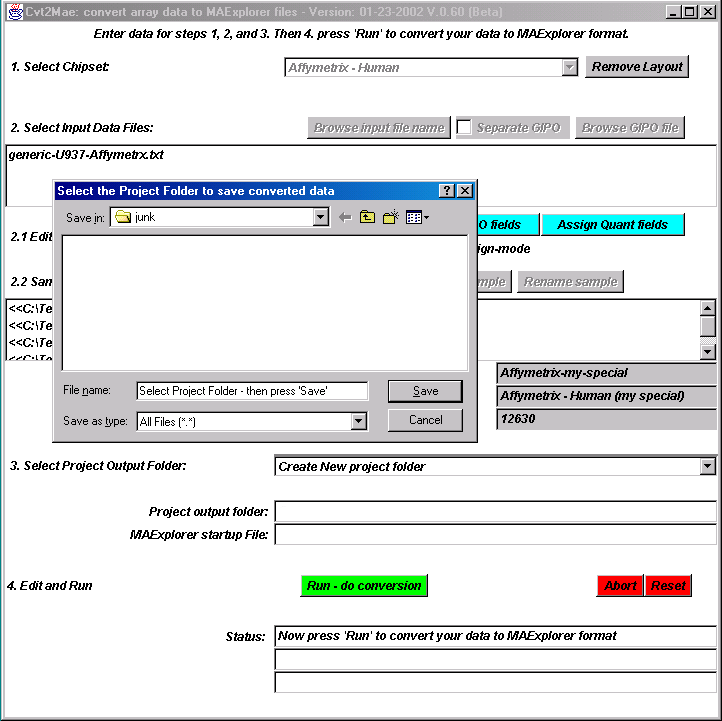
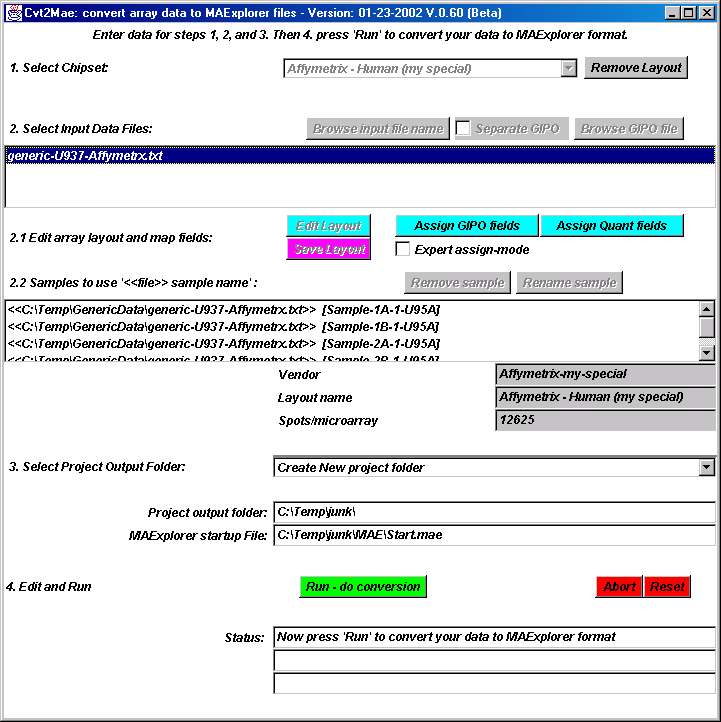
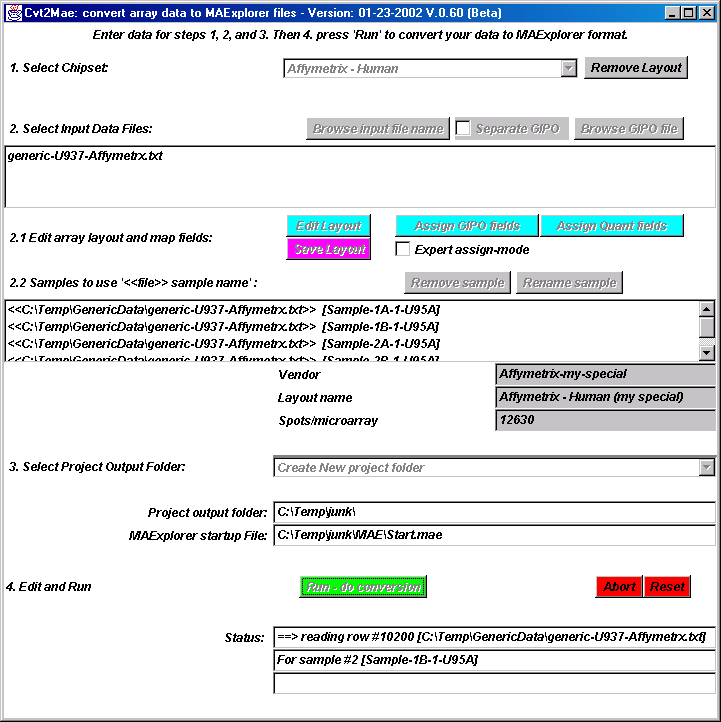
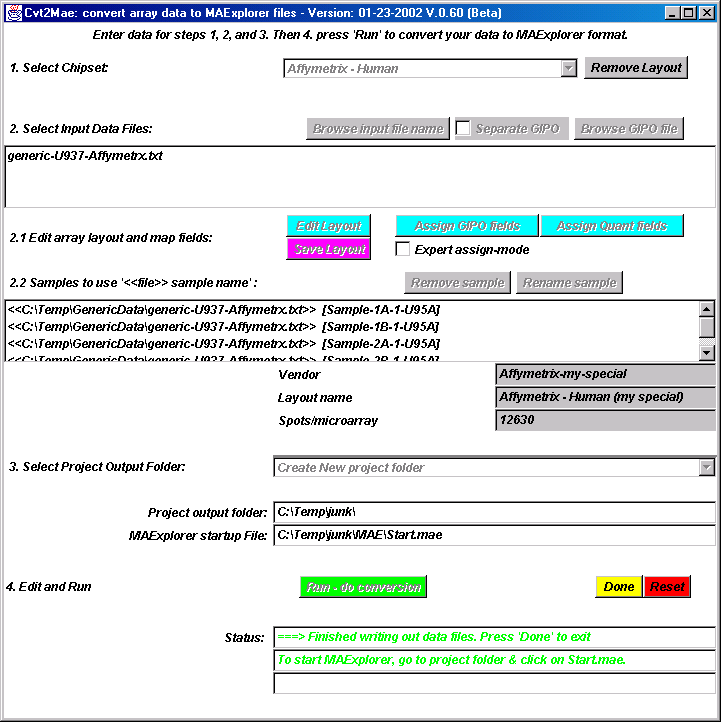

{kind=link}
{kind=link}
{kind=link}