
Figure 1. Overview of MAExplorer exploratory data analysis
system. Initial data preparation steps are performed prior
to analysis by MAExplorer and are indicated by cyan
italics at the top of the figure. The primary data consists of
quantified microarray image data as well as corresponding qualitative
clone ID, gene-in-plate-order (GIPO or print-table, etc.), gene name,
hypertext base references and related information. After the
microarrays are hybridized, they are scanned and spots quantified
using image spot quantification programs. These lists are then saved
for each array in a tab-delimited file. Microarray image
quantification may be performed by various software such as Axon's
GenePix(TM), Scanalyze, Molecular Dynamics
ImageQuant(TM), Research Genetics' Pathways(TM),
etc. When used as a stand-alone application, data may be saved on the
local computer for local off-line use, and direct access to other
Internet genomic databases may be made without using a proxy server.
[DEPRICATED: When used as an applet, this auxiliary
databases and the MAExplorer Jar files are copied to the Web server or
local file system (in the case of the stand-alone version) where they
are then available to be downloaded by users. When a user invokes a
Web page containing the Java applet, it first downloads the applet
that then downloads auxiliary databases including a configuration file
that describes the array data. It then downloads the subset of
quantified microarray spot data files requested for the set of
hybridized samples being investigated. Additional samples may be
downloaded at any time. When the user selects an operation that
requires access to Web databases not residing on the MAExplorer Web
server, implicit Java security restrictions prevent the applet from
going directly to these other Web servers. Instead, it requests the
MAExplorer proxy server request the data from the foreign Web server,
and then returns it back to the user's Web browser. ]
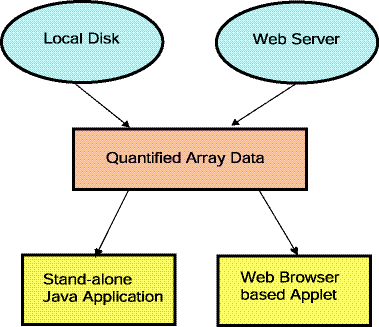
Figure 1.1.1 Overview of MAExplorer exploratory data analysis
system. MAExplorer is used as a stand-alone application on local
data. [Its use as a Web browser applet has been DEPRICATED. In
the case of the applet, it may only access quantified array data from
the Web server that launched the applet.]
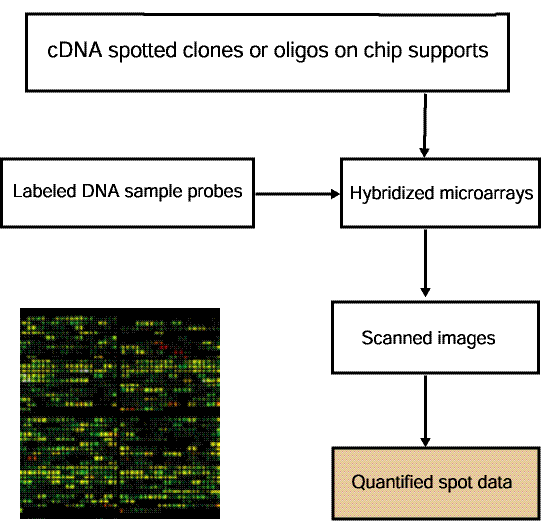
Figure 1.1.2 Overview of data preparation for quantified spot data
used by MAExplorer. MAExplorer handles quantified spot data as
shown in this figure. Arrays are hybridized against labeled samples
are scanned and spots are quantified into spot data files. Quantified
spot data is represented as tab-delimited data with data for one
spot/row. Each spot is identified in this file by its grid
coordinates (grid, grid row, grid column) with image (X,Y) coordinates
being optional. Quantified spot data includes the raw spot intensity
for each channel (in the case of multiple channels such as Cy3, Cy5,
etc.). If the original data has background spot intensity values, then
that may be included as well - otherwise no background data will be
available for background correction. The spot data is discussed in
more detail in Section 1.1 and Appendix C.1, and Appendix C.3.
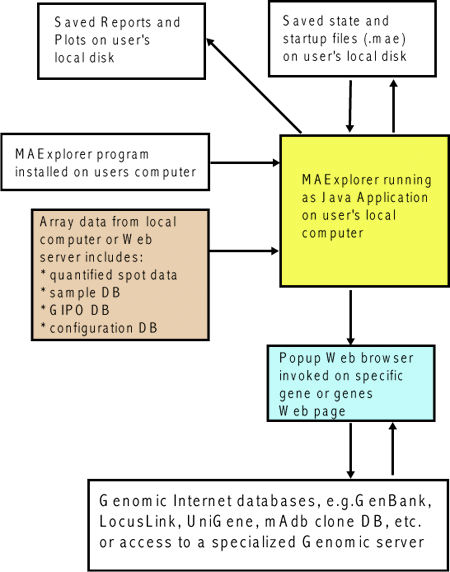
Figure 1.1.3 Overview of running MAExplorer as a stand-alone
application. The preferred way of running MAExplorer is as a
stand-alone application. There are distinct advantages in running
MAExplorer as an application in that data and the exploration state
may be saved on the users local computer, direct access to genomic
servers is easier (no proxy server required - see Figure 1.4). MAExplorer plugin
extensions (MAEPlugins) may only be used with the stand-alone
version. Since MAExplorer is packaged for
download for a variety of operating systems, using this method is
not difficult to set up and the MAEPlugins should run on a variety of
operating systems.
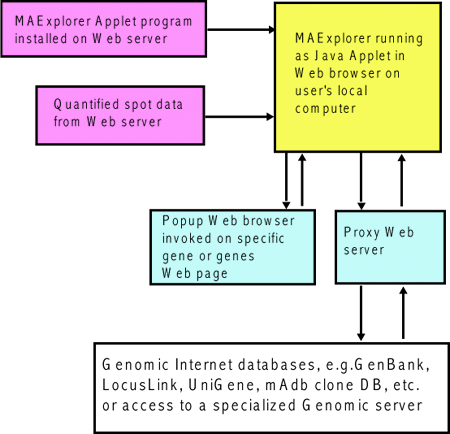
Figure 1.1.4 [DEPRICATED] Overview of running MAExplorer as a Web
browser applet. An alternative way of running MAExplorer on
existing databases is as a Web-browser applet. There advantage of this
method is that no software installation is required on the user's
computer. However, the user may not save data and the exploration
state on their local computer. Furthermore, direct access to genomic
servers requires a proxy server. MAExplorer plugin extensions
(MAEPlugins) may not be used with the the applet version. The Mammary Genome Anatomy Program
(MGAP) originally used the MAExplorer applet.
In MAExplorer we refer to grids by letter names (A,B,C,...) and fields
by F1 and F2. If you are using Cy3/Cy5 ratio data and the Cy3 and Cy5
data is available as independent channels for each HP sample, then
operations that use F1 and F2 will use the Cy3 and Cy5 data for
various operations such as scatter plots (Cy3 vs Cy5), etc. If there
is only one field in an array (i.e. no duplicate grids), then when
MAExplorer is run, operations and menus describing F1 and F2
operations will not be available.
Using duplicate (F1 and F2) spots allows us to get an estimate of the
hybridization variance within an array and is used to compute the
(F1,F2) gene coefficient
of variation (CV) used in the gene data Filter to remove noisy
data before looking for additional differences. Note that if Cy3/Cy5
data is used, then F1 and F2 duplicates are not allowed as MAExplorer
uses the (F1,F2) data to hold the(Cy3,Cy5) data for a hybridized
sample.
Example of a MAExplorer database -
http://www.lecb.ncifcrf.gov/mae - the public MGAP DB
The Mammary Genome Anatomy Program (MGAP) microarrays of cDNA clones
from mouse mammary tissue (collaboration with Research Genetics) were
hybridized with 33P radio-labeled samples. These were then
used to charge fluorescing plates. See the MGAP site for more
documentation on the database and preparation procedures. The
hybridized arrays are scanned on a phospho-imager scanner at high
resolution. Spot data was quantified from these images using the
Research Genetics' "Pathways 2.01" program which generated
tab-delimited data files. This data also includes the microarray grid
point locations (field, grid, grid row, grid col) from the associated
microarray description data files (grid-in-plate-order data). When you
download MAExplorer, you will also
download the public MGAP dataset.1.1 Microarrays and notation used with MAExplorer
In general, microarrays are hybridized using cDNA samples derived from
mRNA labeled with either radio-label, biotin, fluorescent dyes, or
other methods (see Schulze,
2001) for review of the technology). MAExplorer may be used to
construct databases using single-labeled sample intensity (e.g.,
Affymetrix, 33P radio-labeled, etc.) and double-labeled
ratio fluorescent (i.e. Cy3/Cy5) data arrays with different GIPO
geometries.
Definition of "Condition list of samples"
Samples are organized into Condition Lists of samples (generally
replicate samples). These may be used in various statistical and
clustering tests. There are three built-in lists of samples called
the HP-X 'set', the HP-Y 'set' and the HP-E list. The X and Y sets are
used in various 2 condition tests such as the t-Test between the X and Y
sets (Section 2.4.3). The HP-E list is an ordered expression list
of samples used in clustering and in displaying expression
profiles. You may interactively define new or edit
named condition lists using a graphical wizard (Section 2.6), manipulate and assign them to the HP-X
'set', HP-Y 'set' and HP-E list. Some examples of condition lists
might be (assuming you have the data available in your database):
Virgin = ( V.1, V.2, V.3 )
Pregnacy = ( P13.1, P13.2, P13.3 )
Lactation = ( L3.1, L3.2, L3.3 )
Involution = ( I4.1, I4.2, I4.3 )
Definition of "Ordered Condition list" of multiple condition lists
We further extend this paradigm by defining a meta-data structure
called the "Ordered Condition List" or OCL. This is an list of
multiple conditions that you have previously defined. The OCL
may be sorted if you want and the data lends itself to
sorting. E.g., a time series of conditions lends itself to sorting -
different types of diagnoses may not. The OCL may be used in various
statistical tests (e.g., the F-test applied to the current OCL - see Section 2.4.3)). You may interactively
define new or
edit named Ordered Condition Lists using a graphical wizard
(Section 2.7).An example of an ordered condition list might be:
Partuition= ( Virgin, Pregnacy, Lactation, Involution )
Definition of "intensity" for single-labeled samples
MAExplorer uses the term "intensity" in slightly different ways
dependent on whether you are using the single-labeled or fluorescent
double-labeled data. For single-labeled data, "intensity" is the raw
quantified data value as measured by the image scanner. Raw data must
be normalized between samples in order to compare it between
samples. Therefore, to compare N samples, you must first normalize the data and then
compare them.Definition of "intensity" for fluorescent double-labeled samples
For fluorescent double-labeled data, the Cy3 and Cy5 dye-labeled (for
example) measurements are the raw quantified data values as measured
by the image scanner. In this case, "intensity" is defined as the
ratio of Cy3 to Cy5 (i.e. Cy3/Cy5). If you wish to look at the ratio
as Cy5/Cy3, you may flip the two channels on a per-sample basis (see
Section 2.2.2 for more
details).Issues of experimental design of microarray experiments
Some of the issues involved in experimental design
(setting up experiments) based on the types of arrays are discussed in
Section 3.1.1 for (Cy3/Cy5)-labeled as well as 33P-labeled
samples. Poorly designed experiments will not yield significant
statistical results, so attention should be paid to developing an
adequate and robust design for your data given costs of doing
experiments as well as statistical constraints on analyzing the data.
Actual and "Pseudoarray" image geometry
The main MAExplorer windows contains a pseudoarray image for
visualization purposes. It may or may not correspond the spot
positions on the actual array. This array geometry is defined by the
number of replicate Fields (normally 1) each of which contains a
number of grids (also called "blocks") containing a number of
rows/grid and columns/grid of spots. If there is no explicit array
geometry or spot (X,Y) coordinate data available but simply gene
identifiers and intensity data, then an arbitrary pseudoarray geometry
is generated. If there is an explicit array geometry, then it waill
draw the pseudoarray using this geometry. The database configuration
determines which method will be used and is discussed in Appendix C.5. If there is no
explicit grid geometry, the number of spot Locations (e.g., IncyteID,
Affymetrix probe_set) may be used to synthesize a set of grids of a
size that is reasonable for viewing with MAExplorer. This is done in
the Cvt2Mae array data
conversion program when the array
geometry (#grids, #rows/grid, #columns/grid) is not known. This
conversion is not done in MAExplorer itself.
In Cvt2Mae we generate a visually appealing pseudoarray image geometry
if no array geometry is specified with the data (e.g. Affymetrix data,
etc). It maps the number of N spot data entries to a
(#grids,#grid-rows,#grid-columns). The algorithm is given in Appendix C.6 as well as
a suggestion for handling
non-standard geometries using Cvt2Mae.
Gene coordinate numbering on the microarray
A gene coordinate
numbering is a mapping of gene identifiers to locations on the
array for a particular array geometry. These are described by
grids (or blocks), each consisting of grid rows by
grid columns of spots. The grids may be repeated on the array
and constitute duplicate fields. Some arrays group subsets of
grids into meta-grids which are specified by meta-grid
rows by meta-grid columns of grids. MAExplorer can handle
grids but not meta-grids. In the case where there is no
array grid geometry specified or meta-grids are used, an arbitrary
pseudoarray geometry can be constructed to serve as a basis to display
the microarray pseudoimage (see the Algorithm for constructing the
pseudo array from a list of spots in Appendix C.6).




{kind=link}